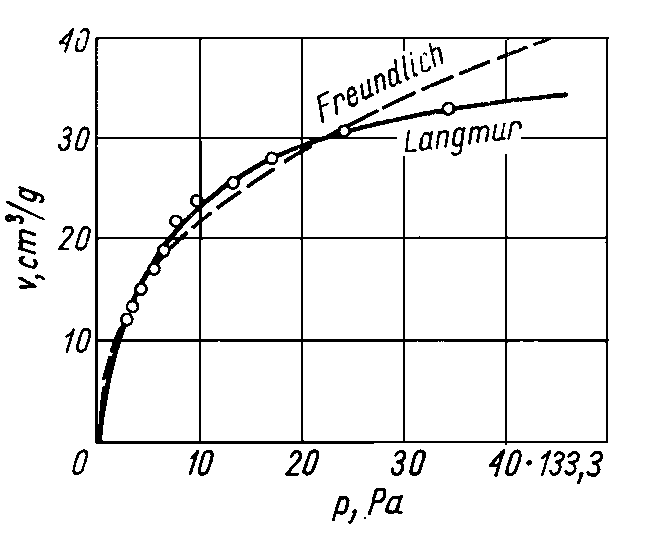
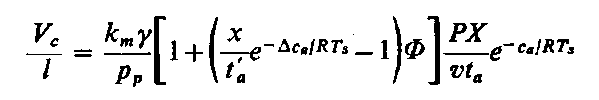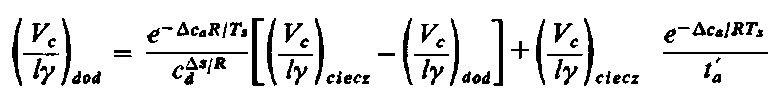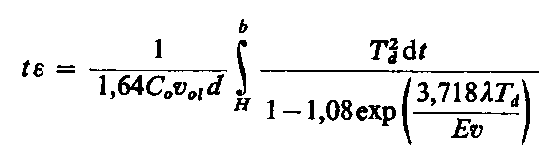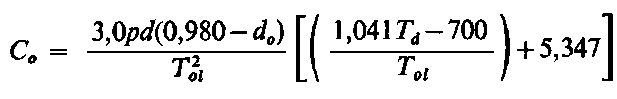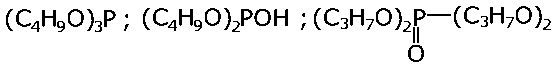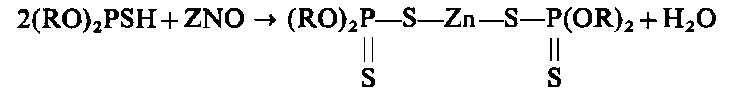| |
5.3. Smarność
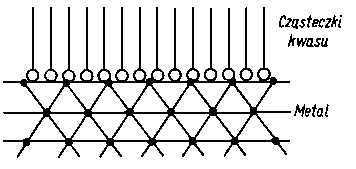
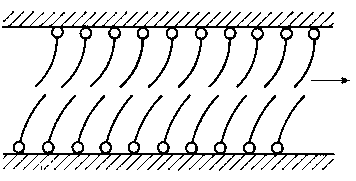
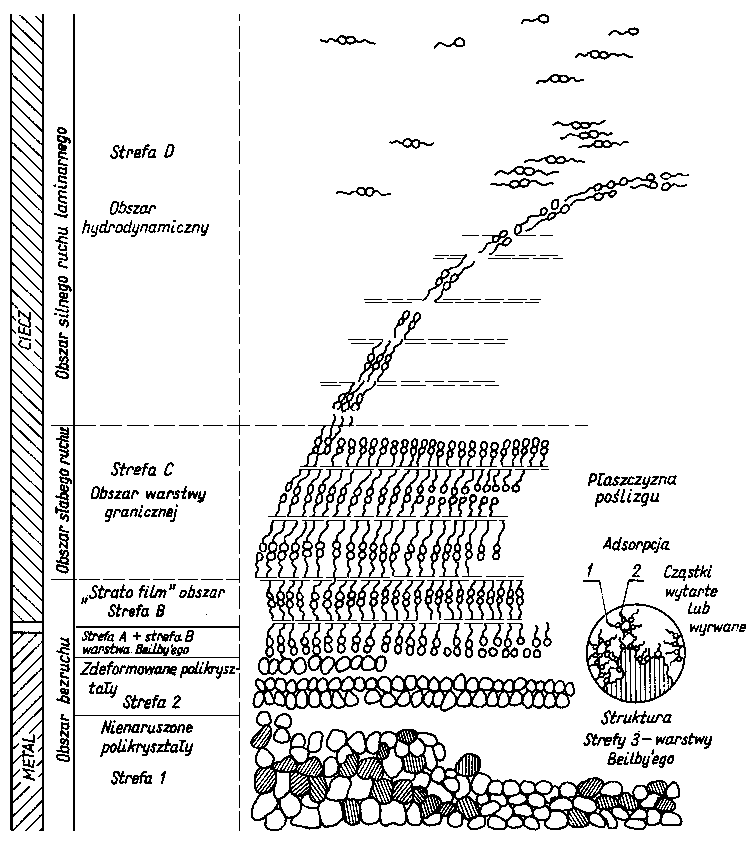
Smarność jest własnością substancji smarującej charakteryzującą jej zachowanie w warunkach tarcia granicznego. Określa ona zdolność do wytworzenia trwałej warstwy granicznej w wyniku adsorpcji (chemisorpcji) na ciałach stałych (podłożu). Podatność ta jest różna w stosunku do różnych rodzajów ciał stałych, z którymi kontaktuje substancja smarująca. Miarą smarności jest trwałość warstwy granicznej, a więc trwałość związania substancji smarującej z podłożem. Trwałość tę można określać zarówno w czasie tworzenia warstwy granicznej (z efektów towarzyszących, np. z ciepła sorpcji) lub też w czasie jego niszczenia (np. z ilości energii, jaką trzeba włożyć aby przerwać warstwę). O trwałości warstwy granicznej można również wnioskować pośrednio ze zjawisk związanych ze smarnością, jak procesów zużycia, skłonności do zacierania itp. (podejście diagnostyczne). Smarność jako podatność do adsorpcji można badać zarówno w warunkach statycznych, jak i dynamicznych.
Pojęcie smarności (lubricity) pojawiło się po raz pierwszy w ostatnich latach ubiegłego stulecia. Pojęcie to wprowadził Kingsburry (14) stwierdzając, że przy stosowaniu olejów o zbliżonych wartościach lepkości istnieją znaczne różnice w oporach tarcia.
Hardy wprowadził nowy termin określający zdolność olejów do tworzenia warstwy granicznej, tzw. oleistość (oiliness). Definicja smarności przyjęta przez SAE (Society of Automative Engineers) jest następująca: smarność jest miarą różnicy oporów tarcia, gdy porównuje się własności różnych smarów o tych samych lepkościach w tych samych warunkach (67).
Podobną definicję przyjęto na Konferencji Smarowniczej w Londynie (68), gdzie zdefiniowano smarność jako ... przyczynę różnicy wartości współczynników tarcia przy zastosowaniu dwóch smarów o tej samej lepkości, tej samej temperaturze i w identycznych warunkach pomiaru". Definicja ta jest używana często, pomimo jej oczywistej nieprecyzyjności.
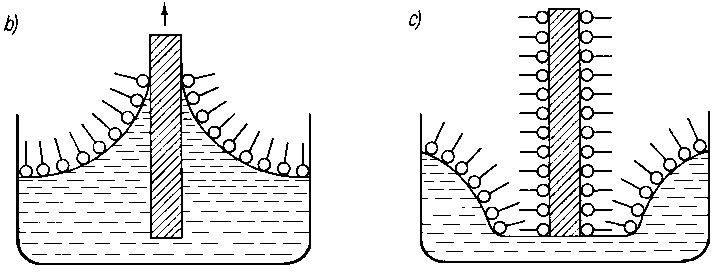
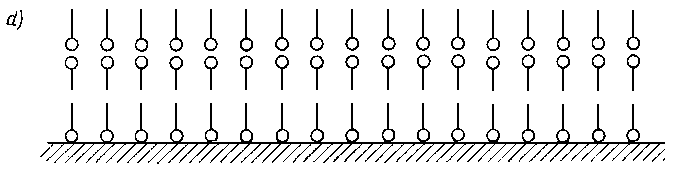
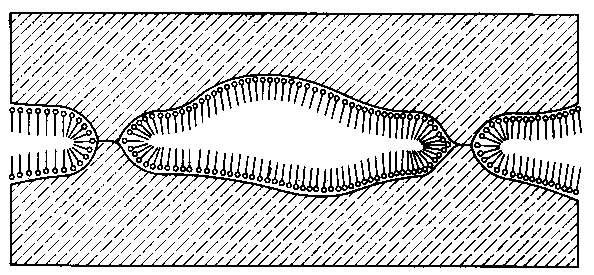
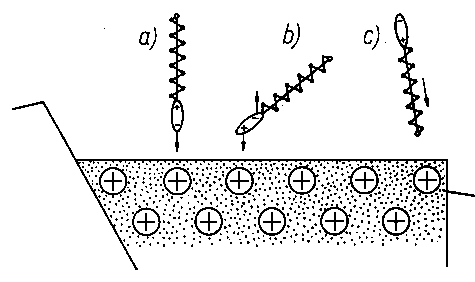
A. Wachal dokonał analizy poglądów różnych autorów dotyczącej zjawiska smarności (69) stwierdzając, że istnieje ogromne zróżnicowanie w tym zakresie. Tak na przykład wielu autorów uważa, że oprócz zmniejszenia wartości współczynnika tarcia przy smarowaniu granicznym smarność polepsza się wraz ze zwiększeniem zwilżalności metalu przez olej. Uzależnia się ją również od wartości napięcia powierzchniowego na granicy faz olej-metal lub olej-powietrze oraz od kąta zwilżenia metalu przez olej. Niektórzy autorzy uważają, że smarnością odznaczają się wyłącznie takie substancje, które zwilżają daną powierzchnię i że smarność jest ściśle uzależniona od wartości napięcia powierzchniowego. Dlatego też staje się zrozumiałe to, że zjawiska napięcia powierzchniowego (70) uważano za główną przyczynę występowania własności smarnych cieczy. Nie ulega wątpliwości, że zjawisko kapilarności, a głównie kształt menisku, jest jednym z głównych parametrów efektu zwilżania, gdyż siły kapilarne pozwalają substancji smarującej przenikać w największe nierówności między stykającymi się powierzchniami.
Z drugiej strony, jak wykazały doświadczenia, najlepszymi własnościami smarnymi odznaczają się oleje o najmniejszej wartości napięcia powierzchniowego i na odwrót ciecze, chociażby o dużej lepkości, ale charakteryzujące się dużą wartością napięcia powierzchniowego, są mało lub zupełnie nie przydatne jako ciecze smarne, jak np. oleje antracenowe, melasa, ługi sulfitowe itp.
Wielu autorów uważało, że przez oznaczenie wartości napięcia powierzchniowego danego oleju przy zetknięciu z określonym metalem można będzie określić przydatność oleju, jako cieczy smarującej dla danej powierzchni. W celu sprawdzenia prawdziwości tego poglądu, wiążącego smarność z kątem zwilżenia i napięciem powierzchniowym, przeprowadzono eksperymentalne sprawdzenie tych poglądów (71). Badania wykazały, że własności te nie są związane bezpośrednio ze smarnością.
Inni autorzy utożsamiają smarność z przyleganiem lub przyczepnością cieczy do powierzchni metalu. Nie jest to określenie ścisłe, gdyż wiele cieczy dobrze przylega do metalu, a nie koniecznie musi charakteryzować się dobrą smarnością.
Kilku autorów sugeruje wnioskowanie o własnościach smarnościowych oleju z wartości ciepła zwilżania lub adsorpcji. E. Piłatowa (72) wprowadziła do oceny smarności tzw. zdolność ochronną oleju. Według tej metody ściśle określoną ilość badanego oleju miesza się z określoną ilością sproszkowanego żelaza (ferrum reductum), a następnie w ciągu określonego czasu poddaje się tę mieszaninę działaniu kwasu siarkowego i oznacza ilość żelaza rozpuszczonego w kwasie, tj. nie chronionego przez olej. Piłatowa uważa, że przy dużej skłonności do adsorpcji olej będzie dobrze chronił powierzchnię metalowego pyłu przed działaniem kwasu.
Oryginalne sformułowanie smarności podaje Fuks (73). Określa on smarność jako „zdolność cieczy do wywoływania małego oporu stycznego kontaktujących się powierzchni ciał stałych, a wysokiego oporu stawianego zbliżania ich pod działania obciążenia normalnego”.
Jeszcze inaczej do zagadnienia smarności podchodzi A. Balada (74). Pisze on: „lepkość myli się czasem z tzw. smarnością, tj. zdolnością do zapewnienia jak najmniejszego tarcia i najmniejszego zużycia oraz do zapobieżenia uszkodzeniom trących się powierzchni”. Praktycy w dziedzinie techniki smarowniczej twierdzą czasem, że potrafią określić smarność przez roztarcie oleju między palcami i obserwacje, jak do nich przylega, a także na podstawie smaku i woni. Taka próba jest oczywiście całkowicie subiektywna i błędna.
Smarność oleju niektórzy mylą nie tylko_z lepkością, ale również ze zwyczajną przyczepnością.
Znacznie bardziej ścisłe sformułowanie podaje K. Kachlik (75). Twierdzi on:
„Smarność oleju nie ma ani definicji, ani jednostki, a jest jedynie jednym z najważniejszych czynników decydujących o dobrej pracy nowoczesnych olejów. Smarność zależy często nie tylko od własności samego oleju, lecz również od własności fizykochemicznych smarowanych powierzchni. Smarność jest tą właściwością smarów, która powoduje różnicę współczynnika tarcia, gdy różne oleje (smary) o tej samej lepkości smarują identyczne łożysko w identycznych warunkach temperatury i obciążenia.
Jak widać z powyższych rozważań, zużycie energii do poruszania urządzeń mechanicznych zależy nie tylko od lepkości oleju, lecz również od smarności.
Smarność zależy wyłącznie od charakteru smaru, czyli od jego zdolności do zobojętniania wolnych sił międzycząsteczkowych na powierzchniach trących metali oraz od rodzaju tych metali. Smarność nie ma nic wspólnego z lepkością oleju, lecz zależy od obecności cząsteczek spolaryzowanych w oleju, które zobojętniają działanie przyciągające wolnych sił międzycząsteczkowych powierzchni trących".
Istnieje również tendencja do nazywania smarnością wszystkich zjawisk występujących w obszarze tarcia wraz z metalami biorącymi udział w procesie tarcia. Przykładem mogą tu być nieodosobnione poglądy Dębickiego (76). Według niego: „Pojęcie smarności nie ma jeszcze jednoznacznej wyczerpującej definicji naukowej. Intuicyjne wyczucie skłania do następującej propozycji definicji jakościowej smarności. Smarność jest to zespól takich właściwości współpracujących ze sobą powierzchni w parze trącej wraz z ciałem pośredniczącym (środki smarne doprowadzane z zewnątrz lub wytwarzające się samorzutnie - "in situ"), które w ściśle określonych parametrach pracy zapewniają taki sposób przemieszczania względem siebie powierzchni elementów trących, przy którym występuje minimalna silą tarcia i minimalne zużycie”.
Nie należy się dziwić, że w sytuacji, kiedy prawie każdy z autorów podkłada pod pojęcie smarności inną treść, gdy brak jest ścisłej definicji i jednostek, pojawiły się poglądy, że smarność należy traktować jako własność, której nigdy nie było. Takim właśnie tytułem opatrzyli swój artykuł Grunberg, Hayward i Jamieson (77) w jednym z najpoważniejszych pism naftowych ("Lubricity - the property that never was"). Wyrażają oni pogląd, że smarność należy zaliczyć do takich samych błędnych i staromodnych terminów jak flogiston (wg dawnej chemii pierwiastek ognia znajdujący się we wszystkich ciałach palnych).
Do powyższego poglądu dołączył się S. E. Holmes (78). Stwierdził on, że w angielskim Słowniku Terminów Naftowych hasło „smarność” zostanie opuszczone jako nienaukowe i nie mające precyzyjnego znaczenia. Przedstawione poglądy spotkały się ze zrozumiałymi sprzeciwami (79).
5.3.1. Eksperymentalna weryfikacja smarności
Dotychczasowe poglądy na temat smarności zostały poddane eksperymentalnej weryfikacji w celu oceny ich prawidłowości i przydatności (69) (71). Najczęściej spotykana jest klasyczna definicja zaproponowana przez twórców tego pojęcia, wg której spośród dwóch olejów o takiej samej lepkości ten ma większą smarność, który w identycznych warunkach smarowania powoduje mniejszą wartość współczynnika tarcia. Takie sformułowanie wprowadza wrażenie, że przy stosowaniu oleju o dobrej smarności chodzi przede wszystkim o zmniejszenie wartości współczynnika tarcia. W rzeczywistości chodzi głównie o niedopuszczenie do zatarcia w warunkach tarcia granicznego i o zmniejszenie zużycia współpracujących elementów.
Uporządkowanie w ułożeniu cząsteczek, jakie wywołują w warstwie granicznej cząsteczki polarne, powinno rzeczywiście zmniejszać wartość współczynnika tarcia, ale z kolei istnienie cząsteczek o dużej zdolności wiązania z podłożem (zarówno na zasadzie oddziaływania międzycząsteczkowego, jak i wiązania chemicznego) może powodować zwiększenie wartości współczynnika tarcia.
Badano olej bazowy P-5 czysty i z dodatkami smarnościowymi (80) dodawanymi w takiej ilości, aby stężenie składnika aktywnego wynosiło około 1,2-proc. cięż. Badania przeprowadzono na aparacie KEWAT-4 i AMSLER. Wyniki badań przedstawiono na rys. 5.49, na którym oznaczenia P-5+C1, P-5+Pb i P-5+
+MoS2 odpowiadają olejowi P-5 z zawartością odpowiednio chloroparafiny, naftenianu ołowiu i dwusiarczku molibdenu.
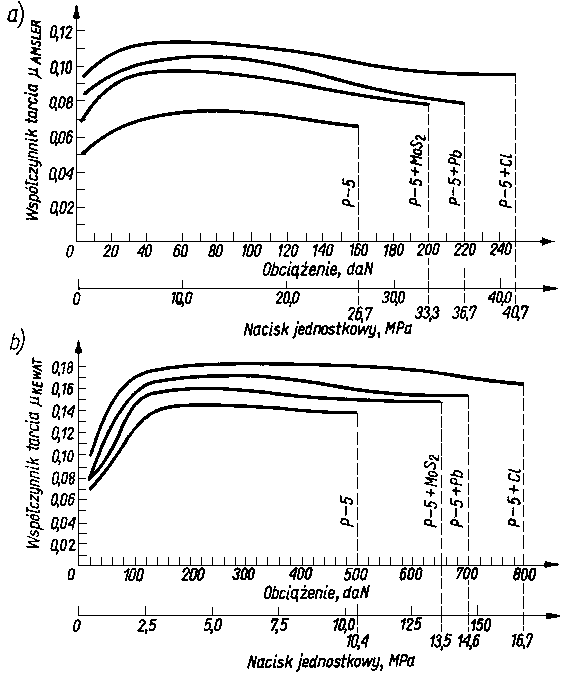 |
Rys. 5.49. Zależność oporów tarcia od nacisków jednostkowych przy smarowaniu olejem bazowym P-5 i olejem z dodatkami podczas badania na aparacie:
- a) Amsler,
- b) KEWAT-4
|
Jak wynika z wykresów, substancje zdolne do chemisorpcji powodują zasadnicze zwiększenie wartości współczynnika tarcia, pomimo że lepkość pozostaje praktycznie nie zmieniona. Oprócz substancji zdolnych do chemisorpcji wprowadzono również do oleju bazowego P-5 olej mineralny o różnym stopniu znitrowania. Przy niskim stopniu znitrowania (równoznaczne z niewielkim momentem dipolowym) wartość współczynnika tarcia zmniejszała się, a przy wysokim stopniu znitrowania (wysoki moment dipolowy) następował wzrost wartości współczynnika tarcia. Olej bazowy czysty miał mniejszą smarność niż ten sam olej z dodatkami smarnościowymi. Należy więc odrzucić klasyczną definicję smarności, jako niezgodną z wynikami badań.
Wprowadzenie dodatków smarnościowych do oleju zwiększa smarność oleju oraz z reguły poprawia jego własności przeciwzużyciowe. Zazwyczaj polepszeniu ulega odporność na jeden lub kilka rodzajów zużycia (np. zużycie ścierne adhezyjne), co zupełnie nie zapobiega równoczesnemu zmniejszeniu odporności na inny rodzaj zużycia, np. zużycie zmęczeniowe (pitting). Stwierdzono to eksperymentalnie w wielu pracach.
Ze względu na to, że ta sama własność substancji smarującej nie może być równocześnie dodatnia i ujemna, nie wydaje się słuszne zastępowanie smarności własnościami przeciwzużyciowymi. Zupełnie poprawne wydaje się natomiast wiązanie smarności z wytrzymałością warstwy granicznej oleju, z tym zastrzeżeniem, że możliwy jest pomiar wytrzymałości warstwy. W aparatach służących głównie do tego celu, jak np. w aparacie czterokulowym przy stosowaniu obowiązującej obecnie metodyki pomiarowej nie mierzy się w zasadzie odporności warstwy oleju na przerwanie, gdyż pojawienie się na kulach obszarów zużycia oznacza, że warstwa oleju musi być cyklicznie przerywana (co powodowało zużywanie) i odnawiana aż do momentu zatarcia. W rzeczywistości mierzy się własności przeciwzużyciowe i przeciwzatarciowe zależne m.in. od smarności, co pozwala wnioskować o tej własności oleju.
Nie budzi zastrzeżeń przyjmowanie jako miary smarności efektów cieplnych adsorpcji, gdyż smarność polega właśnie na wytwarzaniu trwałych warstw granicznych poprzez adsorpcję lub w specjalnych przypadkach poprzez chemisorpcję.
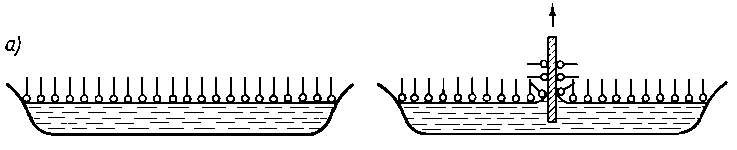
Należało jednak eksperymentalnie sprawdzić poglądy wiążące własności powierzchniowe oleju z jego smarnością. Jak wspomniano, E. Piłatowa (72) opracowała metodę oznaczania zdolności zwilżania powierzchni metalu przez olej. Metoda polega na tym, że znaną (zawsze tę samą) odważkę pyłu żelaza (ferrum reductum) miesza się ze znaną ilością badanego oleju. Gęstą zawiesinę pyłu żelaza w oleju kontaktuje się przez ściśle określony czas z kwasem siarkowym, przy intensywnym mieszaniu gazem obojętnym (np. CO2). Po oddzieleniu kwasu siarkowego od zawiesiny oznacza się w nim zawartość żelaza, którą rozpuścił atakując nie ochronione przez olej powierzchnie metalu. Piłatowa wprowadziła wielkość nazwaną przez nią zdolnością zwilżania Z , określaną wzorem
|
Z = 100 |
( a - b )
a m
| (5.56)
|
gdzie: a - ilość żelaza rozpuszczona w H2SO4 bez obecności oleju, b - ilość żelaza rozpuszczona w H2SO4 po zwilżeniu pyłu żelaza olejem, m - ilość oleju do oznaczenia (np. l gram).
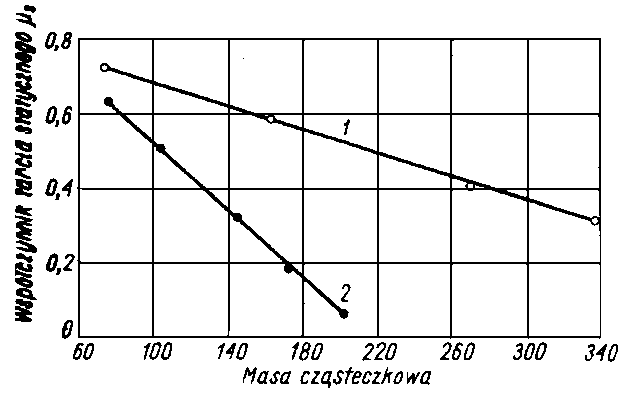
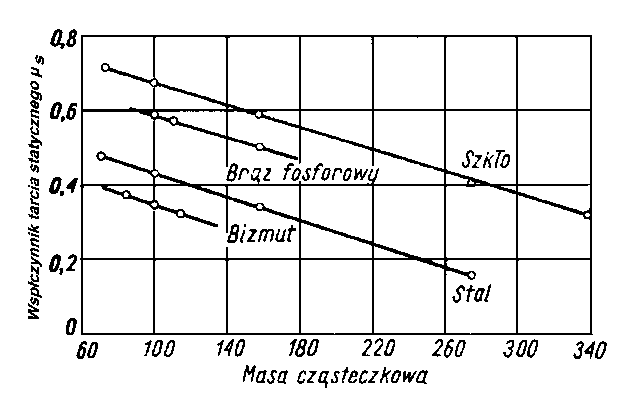
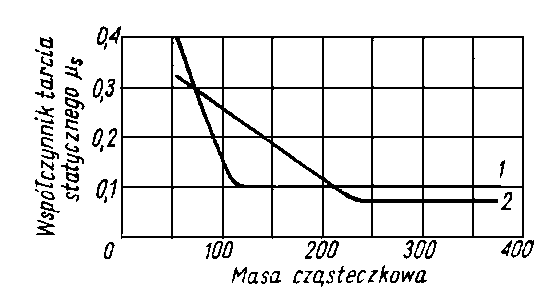
Piłatowa oznaczyła zdolność zwilżania przez wiele olejów. Wielkością tą posługuje się również H. Burstin (40). W tabl. 5.2 przedstawiono przykładowo wartości Z otrzymane przez Piłatową dla kilku olejów, a w tabl. 5.3 - wartości Z wyznaczone przez Burstina.
Oleje parafinowe (np. oleje z ropy pensylwańskiej, parafinum liquidum) mają słabą zdolność zwilżania, natomiast oleje aromatyczne (np. destylaty z ropy uryckiej) i oleje z dużą zawartością żywic wykazują wysoką zdolność zwilżania.
|
Tablica 5.2. Zdolność zwilżania Z przez różne oleje wg Pilatowej (99) | |
Rodzaj oleju | Gęstość (15°C) kg/m³ | Masa cząsteczkowa | Lepkość n
(50 °C)
mm²/s | Wskaźnik lepkości WL | Zdolność zwilżania Z | |
Olej pensylwański | 867,0 | 440 | 27,8 | 102 | 4,8 | |
Olej pensylwański | 891,0 | 710 | 216 | 100 | 5,2 | |
Olej pensylwański cylindrowy | 896,0 | 760 | - | 100 | 6,0 | |
Parafinum liquidum | 888,0 | 408 | 36,4 | 32 | 0,0 | |
Olej aromatyczny l | 1 077,0 | 205 | - | -153 | 17,4 | |
Olej aromatyczny 2 | 1 100,0 | 235 | - | -635 | 13,7 | |
Destylat urycki l | 928,0 | 300 | 25,3 | -214 | 16,6 | |
Destylat urycki 2 | 946,0 | 388 | 140,0 | -49 | 20,5 | |
Destylat urycki 3 | 950,0 | 457 | 300 | 1,5 | 25,0 | |
Destylat urycki 4 | 962,0 | 550 | 1100 | - | 27,6 | |
Olej wrzecionowy +1% GD-162 | - | - | - | - | 31,7 | |
Olej wrzecionowy +0,1% GD-162 | - | - | - | - | 25,4 | |
Olej wrzecionowy +0,01% GD-162 | - | - | - | - | 7,9 | |
Kwasy naftenowe (z nafty) | - | - | - | - | 11,0 | |
Olej wrzecionowy + 5% kwasów naftenowych | - | - | - | - | 0,8
|
|
Tablica 5.3.Zdolność zwilżania Z oleju silnikowego czystego (regular) i z różnymi dodatkami związków polarnych (109) | |
Rodzaj oleju | Z | |
Olej silnikowy (regular) czysty | 21,8 | |
Olej silnikowy + 0,05% oleju siarkowego | 22,1 | |
Olej silnikowy + 0,05% kwasów naftenowych | 26,0 | |
Olej silnikowy + 0,05% estrów metylowych kwasu naftenowego | 26,5 | |
Olej silnikowy + 0,05% stearynianu ołowiu | 27,0 | |
Olej silnikowy + 0,01% utlenionej parafiny | 28,0
|
Przedstawiono również wartości zwilżania przez oleje z dodatkami polarnymi. (Dodatek GD-162 to mieszanina chlorowanych i siarkowych olejów).
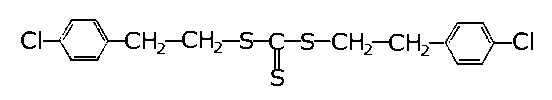
W badaniach sprawdzających (81) stosowano olej P-4 (frakcja z destylacji próżniowej). Do tego oleju wprowadzono jako dodatki polarne zdolne do adsorpcji, ewentualnie do chemisorpcji, czyste związki (alkohol cetylowy CH3(CH2)14CH2OH, kwas palmitynowy CH3(CH2)14COOH i siarka) oraz dodatki handlowe Acorox 88 (dwualkilodwutiofosforan cynku, 50-proc. roztwór w oleju mineralnym), chlorowaną parafinę (ok. 26 atomów węgla i 40% Cl), nitrowany olej parafinowy oraz Dysperbar K (nitrowany Dysperbar 1000-sól barowa kwasów naftosulfonowych).
Tablica 5.4. Własności powierzchniowe i przeciwzatarciowe oleju P-4 z różnymi dodatkami |
Substancja smarująca | Stężenie dodatku % mas. | Stężenie żelaza w roztworze g | Napięcie powierzchniowe kN/m | Zwilżanie
tg ½ q | Średnica śladu zużycia kulek w mm przy obciążeniu P w N | |
stal | miedź | aluminium | 600 | 800 | 1000 | 2000 | 1200 | 1500 | 5000 | 6000 | |
Olej P-4 | 0 | 0,277 9 | 31,2 | 0,145 | 0,171 | 0,084 | 2,34 | 2,65 | 2,71 | 3,06 | zat. | — | |
Olej P-4 + kwas palmitynowy | 0.5 | 0,092 0 | 33,01 | 0,159 | 0,144 | 0,200 | 1,82 | 2,10 | 2,28 | 2,33 | 2,60 | zat. | |
1,5 | 0,132 6 | — | — | — | — | 1,74 | 2,16 | 2,21 | 2,33 | 2,71 | zat. | |
3,0 | 0.164 1 | — | — | — | — | 1,96 | 1,32 | 2,45 | 2,74 | zat. | — | |
Olej P-4 + alkohol cetylowy | 0,5 | 0,133 3 | 33,01 | 0,171 | 0,236 | 0,133 | 1,73 | 2,09 | 2,10 | 2,16 | 2,35 | zat. | |
1,5 | 0,163 1 | 32,01 | 0,169 | 0,222 | 0,179 | 1,77 | 2,10 | 2,40 | 2,76 | zat. | — | |
3,0 | 0,161 8 | 33,01 | 0,163 | 0,232 | 0,174 | 1,76 | 2,21 | 2,38 | 2,43 | zat. | — | |
Olej P-4 + nitrowany olej parafinowy | 0,5 | 0,213 3 | 34,60 | 0,156 | 0,156 | 0,135 | 1,64 | 2,06 | 2,13 | 2,18 | zat. | — | |
1,5 | 0,213 1 | 37,80 | 0,128 | 0,159 | 0,160 | 2,11 | 2,39 | 2,65 | zat. | — | — | |
3,0 | 0,230 6 | 39,40 | 0,097 | 0,146 | 0,225 | 2,06 | 2,24 | 2,39 | 2,51 | zat. | — | |
Olej P-4 + Dysperbar „K” | 0,5 | 0,199 5 | 33,01 | 0,124 | 0,119 | 0,197 | 1,52 | 2,08 | 2,15 | 2,42 | zat. | — | |
1,5 | 0,339 1 | 33,01 | 0,115 | 0,163 | 0,163 | 1,63 | 2,33 | 2,50 | 2,61 | zat. | — | |
3,0 | 0,292 9 | 33,01 | 0,100 | 0,076 | 0,228 | 1,64 | 2,18 | 2,26 | 2,43 | 2,52 | zat. | |
Olej P-4 + Acorox-88 | 0,5 | 0,333 7 | 33,01 | 0,109 | 0,135 | 0,230 | 0,6 | 1,3 | 1,7 | 2,5 | 3,1 | zat. | |
1,5 | 0,367 0 | 34,60 | 0,105 | 0,159 | 0,237 | 0,6 | 1,5 | 2,5 | 2,7 | 3,4 | zat. | |
3,0 | 0,320 5 | 34,60 | 0,094 | 0,131 | 0,221 | 0,6 | 2,2 | 2,5 | 2,9 | 3,4 | zat. | |
Olej P-4 + chloroparafina | 0,5 | 0,130 9 | 33,01 | 0,108 | 0,240 | 0,241 | 0,4 | 1,8 | 2,4 | 2,6 | 3,4 | zat. | |
1,5 | 0,169 3 | 34,60 | 0,116 | 0,216 | 0,208 | 0,4 | 2,2 | 2,6 | 2,8 | 3,6 | zat. | |
3,0 | 0,137 2 | 34,60 | 0,132 | 0,286 | 0,150 | 1,7 | 2,5 | 2,6 | 2,8 | 3,7 | zat. | |
Olej P-4 + siarka | 2,42 | — | 31,22 | 0,111 | 0,214 | 0,095 | 0,6 | 1,09 | 1,1 | 1,42 | 1,81 | 2,01 | 2,38 | zat.
| |
Olej P-4 zawierał wymienione dodatki w stężeniach 0,5; 1,5 i 3,0% mas. Wyjątek stanowiło stężenie siarki 2,92% mas. (stężenie nasycenia).
W przeprowadzonych badaniach nie obliczono zdolności zwilżania Z, lecz podano po prostu ilość rozpuszczonego żelaza (tabl. 5.4 i rys. 5.50).
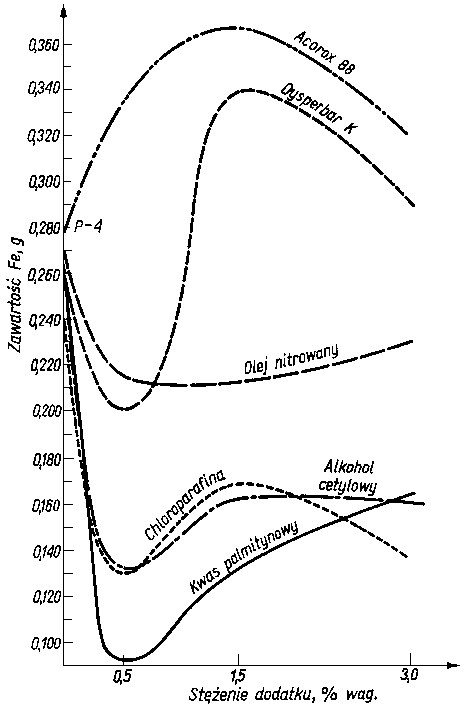 |
Rys. 5.50. Wpływ stężenia różnych dodatków na zdolność ochronną oleju P-4
|
Opierając się na tych samych próbach oleju P-4 czystego i z dodatkami wyznaczono kąt zwilżania przez olej stali, miedzi i aluminium, a ponadto wartość napięcia powierzchniowego oleju. Kąt zwilżania mierzono metodą mikroskopową, rejestrując średnicę i wysokość kropli oleju naniesionej na płytkę metalu, umieszczonej w polu widzenia obiektywu mikroskopu.
Wartość tangensa połowy kąta zwilżania Q obliczano ze wzoru
gdzie: h - wysokość kropli, l - średnica kropli.
Napięcie powierzchniowe oleju oznaczono metodą pęcherzykową. Wyniki pomiarów zostały podane w tabl. 5.4.
Wprowadzenie dodatków smarnościowych do oleju wywołuje na ogół nieznaczny wzrost wartości napięcia powierzchniowego. Większość dodatków polarnych powoduje zwiększenie kąta zwilżania. Wynik pomiaru zależy w dużym stopniu od stanu powierzchni metalu.
Na podstawie analizy wyników badań sprawdzających można stwierdzić, że proponowane przez wielu autorów metody pomiaru własności powierzchniowych olejów w celu oceny własności smarnościowych są do tych celów nieprzydatne i konieczne jest oparcie się na innych metodach oceny smarności.
5.3.2. Pomiary trwałości warstwy granicznej oparte na efektach towarzyszących jej tworzeniu
Graniczna warstwa oleju jest tym trwalsza, im silniej tworzące ją cząsteczki są związane z podłożem. Z kolei im silniejsze tworzy się wiązanie, tym większa ilość energii wydziela się w tym procesie. Mierząc efekty cieplne towarzyszące sorpcji, możemy wnioskować o trwałości powstającej warstwy granicznej.
Do badań wpływu różnych substancji na tworzenie warstwy granicznej zastosowano pomiary ciepła adsorpcji. Pomiary te wykonywano za pomocą mikrokalorymetru przepływowego A. J. Groszka. Schemat komory pomiarowej tego mikrokalorymetru przedstawiono na rys. 5.51.
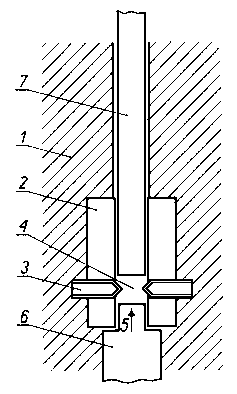 |
Rys. 5.51. Komora pomiarowa mikrokalorymetru;
- - blok metalowy,
- - komora z teflonu (PTFE),
- - termistor,
- - adsorbent,
- - przepuszczalna siatka,
- - rura wylotowa,
- - rura wlotowa
|
Zasada pomiaru jest następująca. Przez sproszkowany adsorbent, którym jest zazwyczaj żelazo lub jego tlenki, umieszczony na przepuszczalnej gazie, przesącza się ze stałą prędkością ok. 0,10 ml/min roztwór substancji zdolnych do sorpcji. Efekt cieplny jaki występuje podczas sorpcji mierzy się za pomocą termistorów i rejestruje na taśmie. Ilość adsorbentu umieszczonego w komorze pomiarowej odmierza się objętościowo (0,10 ¸ 0,12 ml), a następnie waży na wadze analitycznej. Adsorbent powinien być umieszczony w komorze tak, aby termistory były w połowie grubości jego warstwy.
A. J. Groszek (82), (83), (84) mierzył efekt cieplny sorpcji dwiema metodami. Pierwsza polega na przesączaniu przez adsorbent roztworu o znanym stężeniu przez określony czas i mierzeniu całkowitej ilości wydzielonego ciepła.
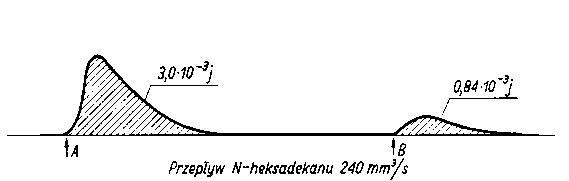 | |
Rys. 5.52. Adsorpcja pulsacyjna kwasu heksadekanowego i kwasu kapronowego z roztworu w heksadekanie;
powierzchnia adsorbenta = 0,27 m²
A - wstrzyknięcie 10 µg kwasu heksadekanowego,
B - wstrzyknięcie 5 µg kwasu kapronowego
|
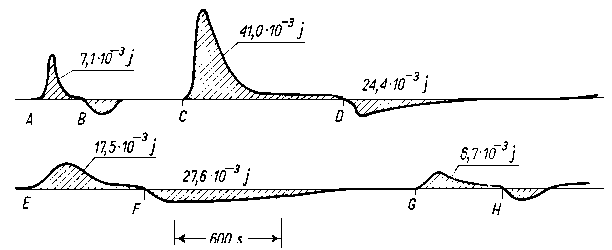 | |
Rys. 5.53. Efekty cieplne towarzyszące adsorpcji: n-oktadekanolu i kwasu n-oktadekanowego z roztworu w n-heptanie, na pyle żelaza;
powierzchnia adsorbenta = 0,15 m²
A - adsorpcja roztworu (2 g/dm³) n - oktadekanolu w n-heptanie,
B - desorpcja n-heptanem,
C - adsorpcja roztworu (2 g/dm³) kwasu oktadekanowego,
D - desorpcja n-heptanem,
E - powtórna adsorpcja roztworu (2 g/dm³) kwasu oktadekanowego,
F - desorpcja n-heptanem,
G - powtórna adsorpcja n-oktadekanolu,
H - desorpcja n-heptanem
|
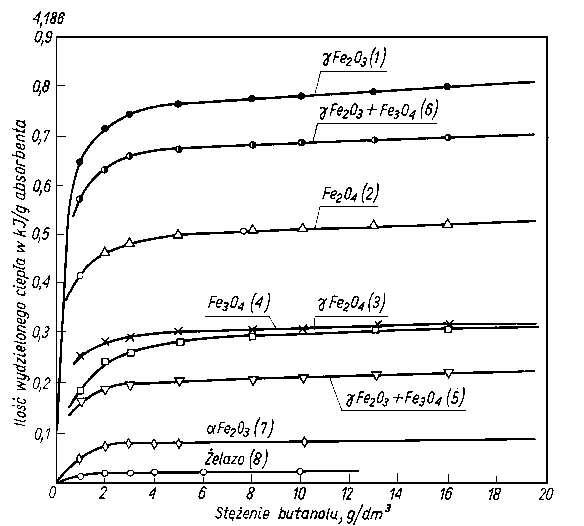 |
Rys. 5.54. Adsorpcja butanolu rozpuszczonego w n-heptanie na żelazie i jego tlenkach (numery krzywych oznaczają rodzaj adsorbenta-objaśnienia w tabl. 5.5)
|
Druga metoda, nazwana przez autora metodą impulsową, polega na przepuszczaniu przez adsorbent jedynie cieczy nośnej. Po zarejestrowaniu na taśmie linii podstawowej wstrzykuje się do cieczy nośnej określoną ilość substancji zdolnej do sorpcji. Jej zaadsorbowanie na powierzchni adsorbenta powoduje wydzielenie określonej ilości ciepła, co jest zarejestrowane na wykresie w postaci wybrzuszenia (piku) (rys. 5.52). W przypadku desorpcji następuje pochłanianie ciepła i pojawia się na wykresie pik ujemny (rys. 5.53).
W celu wyrażenia pola piku w jednostkach ciepła należy przeprowadzić cechowanie mikrokalorymetru. W tym celu wprowadza się do przestrzeni zapełnionej adsorbentem spiralkę grzejną, którą rozgrzewa się impulsami elektrycznymi o znanym natężeniu, rejestrując równocześnie wykres na taśmie rejestratora. Znając ilość ciepła wprowadzoną do układu przez spiralkę grzejną i mając zarejestrowany wykres, możemy obliczyć ilość ciepła, którym odpowiada jednostka powierzchni wykresu.
Wartość efektu cieplnego podczas sorpcji zależy nie tylko od rodzaju sorbującej substancji, ale również i od rodzaju adsorbenta. Na rys. 5.54 przedstawiono zależność ciepła sorpcji butanolu na żelazie i jego tlenkach w zależności od stężenia butanolu w roztworze heptanu (tabl. 5.5).
|
Tablica 5.5. Własności pyłu żelaza i jego tlenków użytych jako adsorbentów (wg A. J. Groszku) | |
Adsorbent nr | Rodzaj adsorbentu (wg oznaczeń opartych na dyfrakcji promieni Roentgena) | Powierzchnia aktywna, m²/g | |
wyznaczona z ciepła adsorpcji butanolu | wyznaczona metodą BET | |
1 | Głównie g-Fe2O3 (0,1 do 1,0% cięż. Al, Cu, Si i Ni) | 21,1 | 19 | |
2 | Głównie Fe3O4 plus małe ilości g-Fe2O3, (ślady Al, Cu) | 17,3 | 13 | |
3 | g-Fe2O3 (ślady Al i Cu) | 11,5 | 12 | |
4 | Fe3O4zawierający < 10% cięż. Mn (ślady Al, Cu, Si) | 13,2 | 9 | |
5 | Mieszanina Fe3O4 i g-Fe2O3 (ślady Al, Si, Pb, Ni i Ti) | 7,5 | 6 | |
6 | Mieszanina Fe3O4 i g-Fe2O3(ślady Ca, Mn, Si, Co) | 20,5 | 22 | |
7 | a-Fe2O3 wysokiej czystości | 3,7 | 3,1 | |
8 | Proszek spektralnie czystego żelaza, mielony w młynie wibracyjnym w heptanie bez dostępu tlenu | 0,7 | » 1,0
|
Z krzywych efektów cieplnych można wnioskować o sposobie ułożenia cząstek na powierzchni adsorbenta. Małe efekty cieplne są spowodowane małą gęstością upakowania i płaskim ułożeniem cząsteczek, natomiast duże efekty cieplne są wynikiem dużej gęstości upakowania przy pionowym ułożeniu cząsteczek.
Adsorpcja różnych związków na powierzchniach metali zmienia w zasadniczy sposób elektryczne przewodnictwo „naskórkowe" metali. Ze względu na to, że pomiary elektryczne są dokładne, szybkie i stosunkowo łatwe w realizacji, badania tego rodzaju mogą być wygodnym źródłem informacji o procesach sorpcyjnych na powierzchniach metali.
5.3.3. Pomiary trwałości warstwy granicznej oparte na efektach towarzyszących jej niszczeniu
5.3.3.1. Zużyciowa interpretacja pomiarów wytrzymałości warstwy
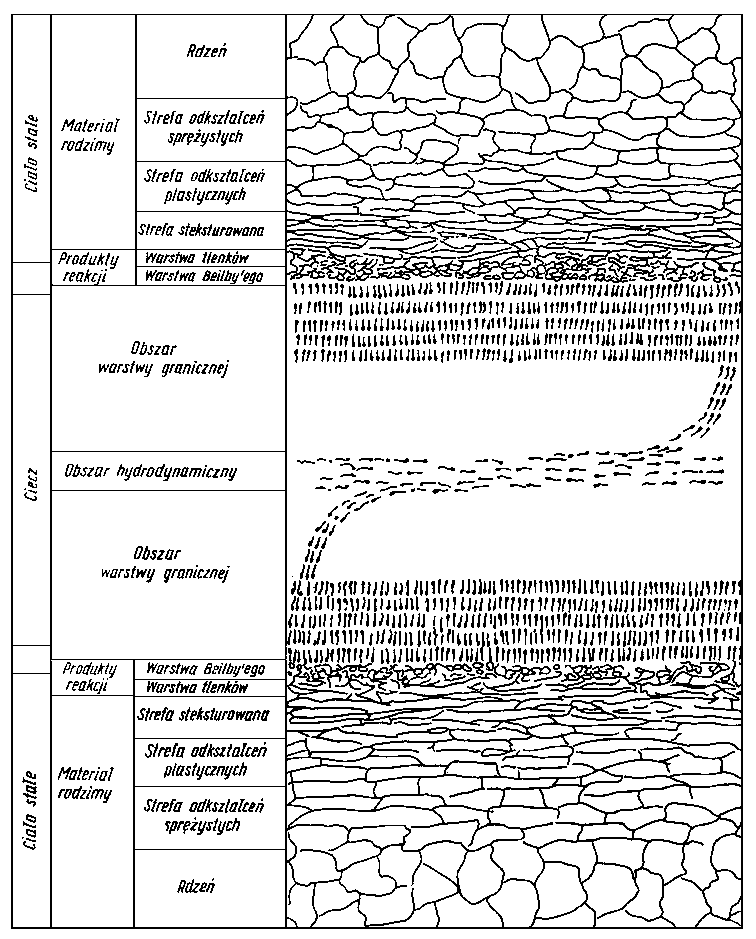
Graniczna warstwa oleju jest dopóty trwała, dopóki jej odporność na przerwanie jest większa lub równa oddziaływaniom niszczącym. Z chwilą przerwania warstwy granicznej następuje zużywanie, a więc oddzielanie cząstek materiału (dekohezja).
W przypadku współpracy skojarzeń, których geometria styku pozwala na zwiększenie powierzchni tarcia w miarę zużywania się elementów (np. styk dwóch kuł, kuli lub trzpienia stożkowego z płaską obracającą się tarczą) po przerwaniu warstwy oleju następuje gwałtowne zużycie tych elementów i wzrost powierzchni tarcia. Przy stałym obciążeniu powoduje to zmniejszenie rzeczywistego nacisku i odbudowę warstwy granicznej oleju.
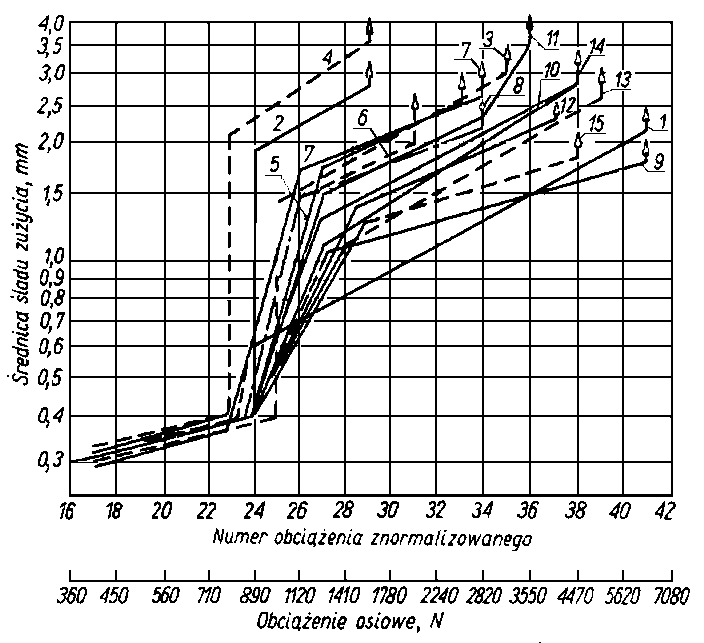
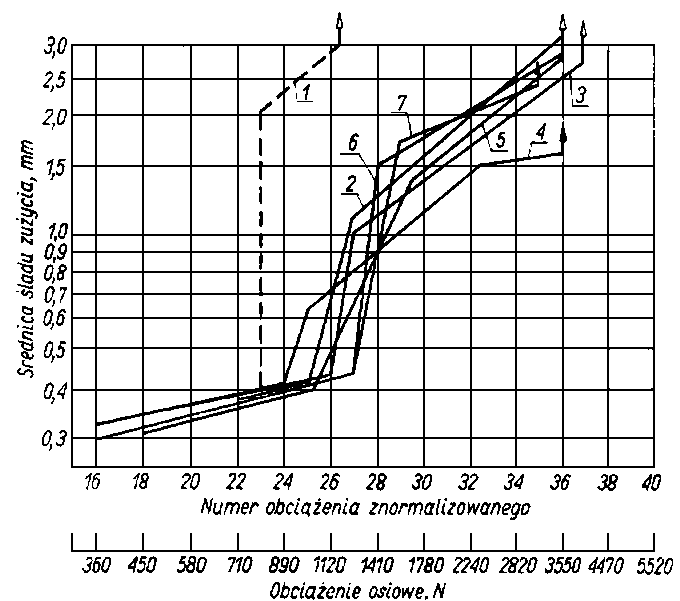
Odporność na zacieranie i zużywanie jest uzależnione od trwałości warstwy granicznej oleju i może być wykorzystywana jako jej miara. Do wyznaczania tak pojętej trwałości warstwy granicznej oleju, a więc smarności, służą różnego rodzaju maszyny tarciowe, np. aparat czterokulowy. Metoda pomiarów za pomocą aparatu czterokulowego w większości krajów została znormalizowana. W metodzie znormalizowanej rozróżnia się następujące rodzaje obciążeń w aparacie czterokuIowym:
- obciążenie nadane P, tj. obciążenie przyłożone do współpracujących elementów;
- obciążenie rzeczywiste kulek Przecz, tj. obciążenie występujące między górną kulą a trzema dolnymi, obliczone wg rozkładu sił w układzie regularnego czworościanu Przecz = 0,408 P;
- obciążenie zespawania Pz tj. największe obciążenie, przy którym w czasie sześćdziesięciosekundowej pracy aparatu następuje zespawanie górnej kuli z zespołem trzech kuł dolnych;
obciążenie właściwe Pwł (zwane również obciążaniem zużycia), tj. obciążenie przypadające na zwiększoną w wyniku zużycia powierzchnię styku kuł
gdzie d- średnica śladu zużycia kuli w mm. przy danym obciążeniu P, 0,52 - współczynnik przeliczeniowy;
- obciążenie skorygowane Pskor obliczone ze wzoru
gdzie: P - obciążenie nadane w N, DH - średnica odkształcenia sprężystego kulki wg Hertza pod danym obciążeniem statycznym obliczona ze wzoru
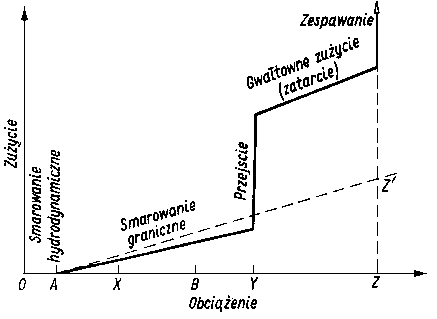
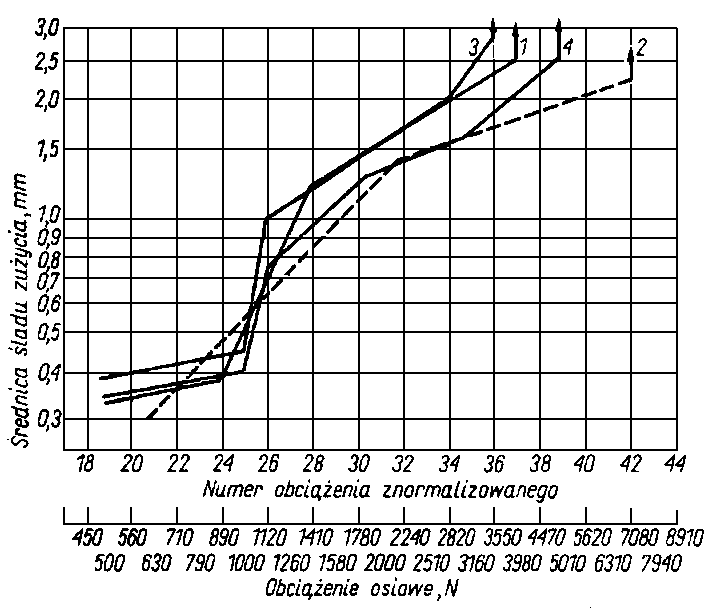
- Przy pomiarach znormalizowanych, prowadzonych za pomocą aparatu czterokulowego, własności przeciwzużyciowe oleju, a więc i pośrednio jego własności smarnościowe, charakteryzuje się wartością średnicy śladu zużycia kuł oraz obciążeniem zespawania (rys. 5.55).
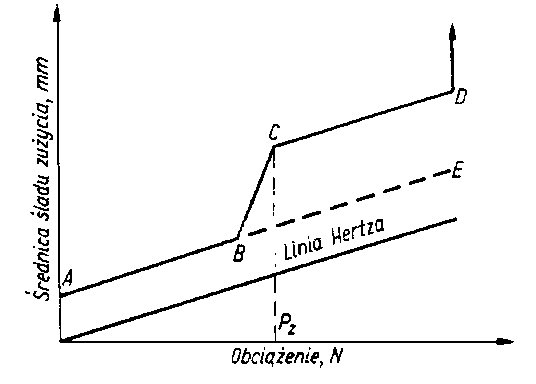 |
Rys. 5.55. Przebieg zużywania elementów tarcia aparatu czterokulowego w funkcji obciążenia;
- ABE - linia kompensacyjna,
- B - największe obciążenie niezacierające,
- BC - początek zacierania,
- CD - intensywne zużywanie,
- D - zespawanie,
- Pz - obciążenie zacierania
|
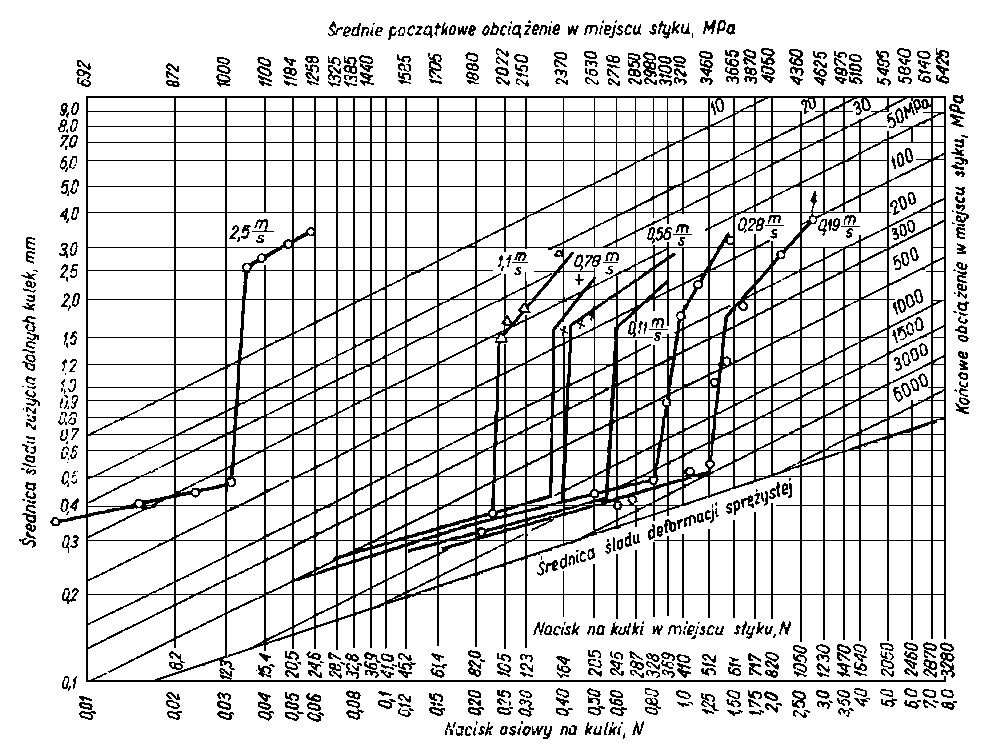 | |
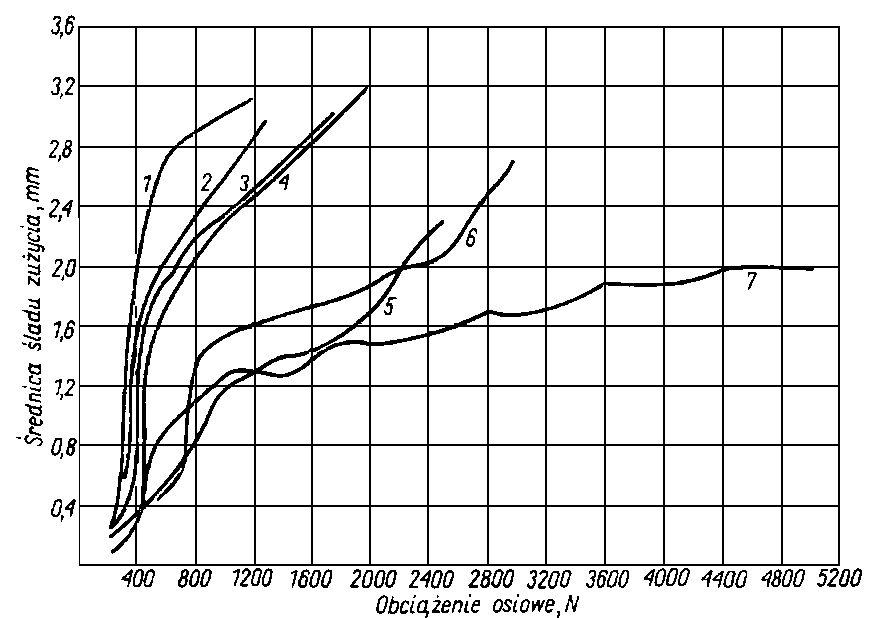
Rys. 5.56. Wpływ prędkości obrotowej na zużywanie kulek w funkcji obciążenia (wg Matwiejewskiego)
|
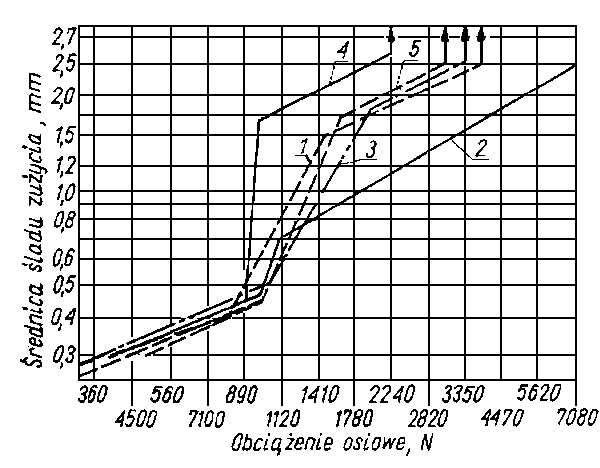
- Miarą smarności może być czas zużywania, czyli czas jaki musi upłynąć przy danym obciążeniu (np. 980 N), aby średnica śladu zużycia osiągnęła wartość l mm (87).
- Za pomocą aparatu czterokulowego można prowadzić wiele innych pomiarów charakteryzujących odporność warstwy granicznej na przerwanie. Jeżeli zaopatrzy się aparat w urządzenie, które umożliwia zwiększanie obciążenia w sposób ciągły, w miarę wydłużania się drogi i czasu tarcia, to powoduje się szczególne spiętrzenie efektów cieplnych w obszarach tarcia, co stwarza szczególnie niesprzyjające
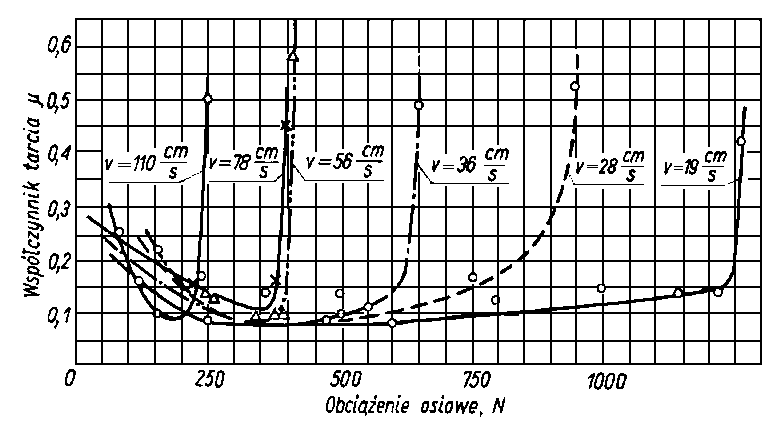 | |
Rys. 5.57. Zależność współczynnika tarcia od obciążenia osiowego przy różnych prędkościach poślizgu (aparat czterokulowy, olej transformatorowy)
|
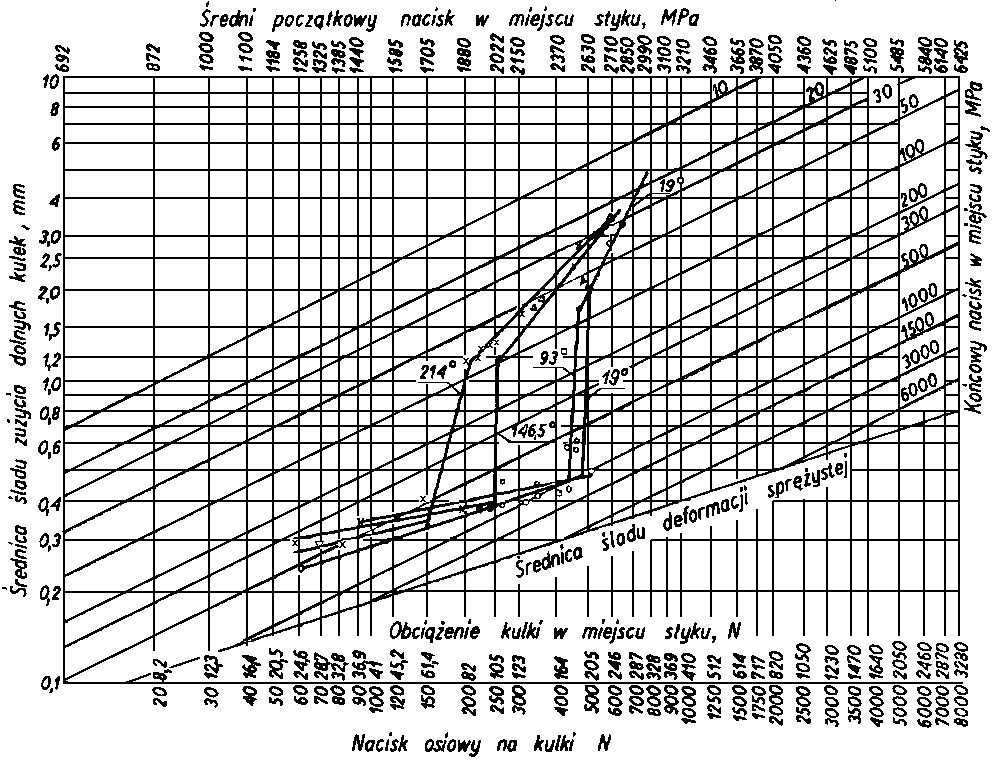 | |
Rys. 5.58. Wpływ temperatury na zużywanie kulek w aparacie czterokulowym (wg Matwiejewskiego)
|
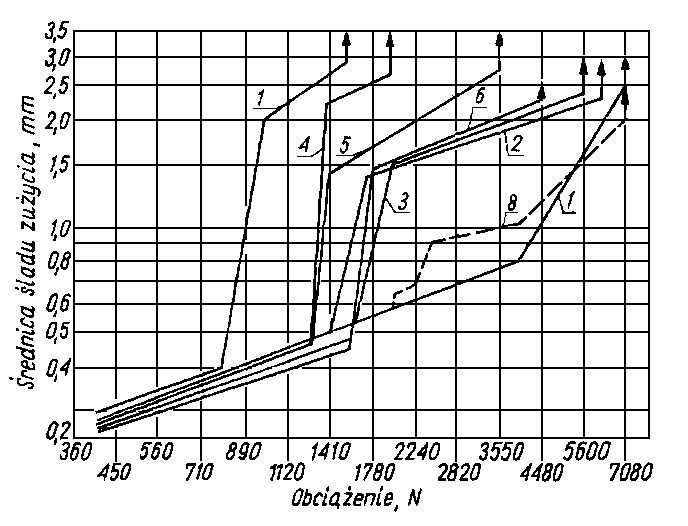
warunki dla pracy oleju. Warunki takie wytrzymują tylko niektóre oleje o dużej smarności. Dodatkowe możliwości oceny oleju uzyskuje się prowadząc pomiary przy zmiennej prędkości obrotowej kulek. Aparat czterokulowy w standardowym wykonaniu ma stałą prędkość obrotową (1450 obr/min). W takim przypadku czynnikiem powodującym niszczenie warstwy granicznej jest wzrost obciążenia i związany z nim wzrost temperatury. Wpływ zmiany prędkości obrotowej na intensywność zużywania kuł przedstawiono na rys. 5.56 i 5.57. Zwłaszcza interesujące są konstrukcyjne wersje aparatu czterokulowego umożliwiające pracę przy bardzo nieznacznych prędkościach obrotowych, np. l obr/min. Zostają wówczas wyeliminowane efekty hydrodynamiczne, a ponadto jest umożliwiony dobry odpływ ciepła.
- Aparat czterokulowy może być również wyposażony w urządzenie do podgrzewania miseczki z kulami lub też do jej chłodzenia. Umożliwia to wykonywanie pomiarów trwałości warstwy granicznej w różnych temperaturach. Wpływ temperatury na zużywanie kuł przedstawiono na rys. 5.58 do 5.60.
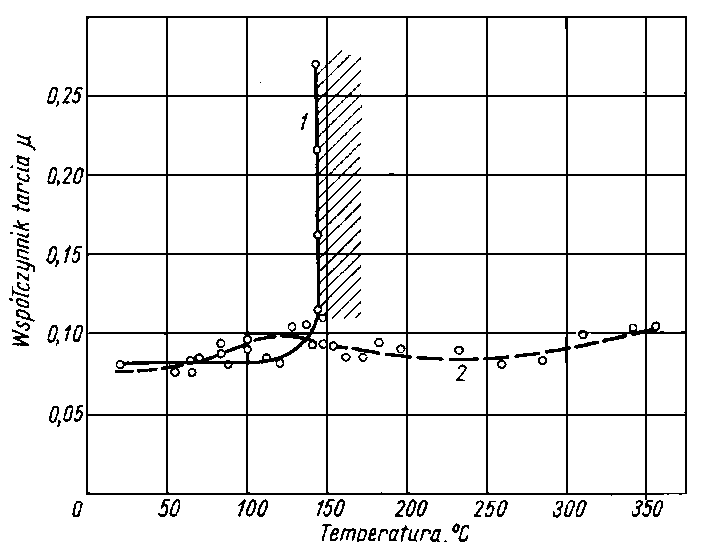 |
Rys. 5.59. Zależność współczynnika tarcia od temperatury (wg Matwiejewskiego);
- v = 0,4 m/s,
- p = 31 500 MPa,
- l - olej SU,
- l - olej SU z dodatkiem siarkowym
|
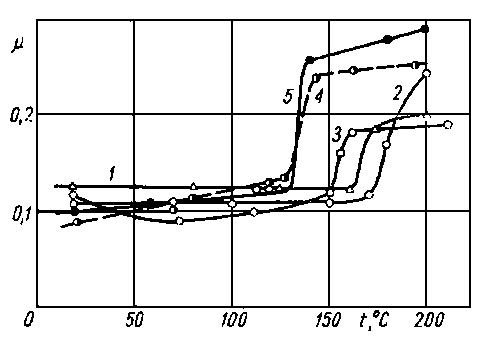 |
Rys. 5.60. Zależność współczynnika tarcia od temperatury dla różnych olejów (wg Matwiejewskiego);
- 1 olej wrzecionowy,
- 2 olej transformatorowy,
- 3 olej silnikowy MS-14,
- 4 brightstock,
- 5 olej silnikowy AK-15
|
- Matwiejewski (92), (93) wprowadził temperaturową ocenę smarności. Pomiar wykonuje się za pomocą czterokulowego aparatu MAST-1 przy bardzo nieznacznej prędkości obrotowej mierząc średnicę zużycia kuł i współczynnik tarcia. Jak widać z rys. 5.58 do 5.60 przy określonej temperaturze występuje gwałtowny wzrost współczynnika tarcia oraz wzrost zużycia kulek, co może służyć do oceny smarności oleju.
- W aparatach czterokulowych wyposażonych w komplety kulek, wykonanych z różnych materiałów (metali i tworzyw polimerowych) możliwa jest ocena wpływu materiałów skojarzonych na twardość warstwy granicznej.
- Blok wprowadził określenie tzw. zwłoki zacierania (88) (seizure delay). Wykorzystał do tego celu wykres przebiegu tarcia, jaki jest zapisywany na prostym rejestratorze, w który jest zaopatrzony każdy standardowy aparat czterokulowy. Zwłoka zacierania jest to czas upływający od początku uruchomienia aparatu aż do wzrostu oporów tarcia (rys. 5.61).
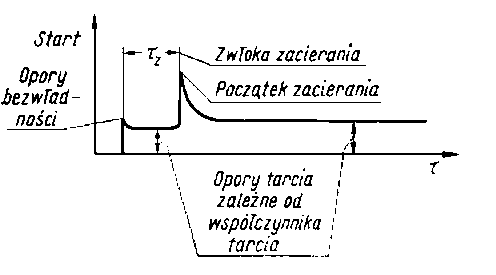 |
Rys. 5.61. Opóźnienie zatarcia (zwłoka zacierania tz ) wg Bloka
|
5.3.3.2. Energetyczna interpretacja pomiarów wytrzymałości warstwy
Dorn (98), (99) ocenia smarność oleju mierząc wartość energii dynamicznej przekształcanej w warstwie granicznej egr (nie uwzględniając przy tym energii wewnętrznej i potencjalnej) wychodząc z podstawowego równania bilansu energetycznego
|
ezasil = ekul + egr
| (5.61)
|
gdzie: ezasil - jednostkowa energia dostarczana do układu (energia zasilania), ekul - jednostkowa energia kulek, egr - jednostkowa energia przekształcana w inne formy energii w warstwie granicznej.
- Energia dynamiczna dostarczana do układu
|
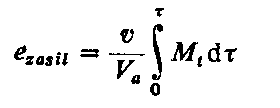
| (5.62)
|
gdzie: v - prędkość obrotowa w m/s, r - promień kulki w mm, t - czas w s. Mt - moment tarcia w daN mm, Va - objętość kulki zaktywizowana w mm³.
- Energia przekształcona w kulkach (energia kulek) ekul
- Można tu wyróżnić energię zużywaną na: oddzielenie materiału, odkształcenia, zmiany w sieci przestrzennej, przemiany fazowe, zmiany struktury, zmiany objętości, podwyższenie temperatury i odprowadzenie ciepła.
- Dla żelaza i stali wartość energii zużywanej na zmiany w sieci przestrzennej jest pomijalnie mała. Dom podaje podstawowe równanie bilansu energetycznego.
|
ezasil = et + ev + ef + |
Qodpr
Va
| (5.63)
|
- Poszczególne człony równania (5.63) składają się z następujących energii:
- - jednostkowa energia topnienia
|
et = 427 d cwł ( tt - tkul )
| (5.64)
|
- - jednostkowa energia zmiany objętości
|
ev = |
g
kv |
( tt - tkul )
| (5.65)
|
- - jednostkowa energia odkształcenia (zmiana formy)
- - jednostkowa energia cieplna odprowadzana
|
Qodpr
Va |
= |
Qprzew + Qkonw + Qprom
Va
| (5.67)
|
gdzie: d - gęstość w kg/m³, cwł - ciepło właściwe w J/(g•°C), tt - temperatura topnienia, tkul - temperatura kulki w °C, g - współczynnik wydłużenia objętościowego 1°C-1, kv - objętościowy współczynnik ściśliwości w mm³/N, kf - jednostkowy opór odkształcenia w MPa, r - jednostkowy logarytmiczny stopień odkształcenia, Qprzew - ciepło przewodzenia w J, Qkonw - ciepło konwekcji w J, Qprom - ciepło promieniowania w J.
- Energia przekształcona w warstwie granicznej
|
egr = echem + edes + ewym
| (5.68)
|
gdzie: echem - energia chemiczna, edes - energia procesów desorpcji i usuwania warstewek powierzchniowych, ewym - energia procesów wymiany.
- W przemianach w warstwie granicznej zużywana jest stosunkowo niewielka ilość energii zasilania, lecz trzeba pamiętać o tym, że poprzez te przemiany dochodzi się do stanu krytycznego, a grubość warstwy granicznej i jej wytrzymałość są decydującymi parametrami obciążalności.
- Weryfikacja eksperymentalna tej teorii polegała na badaniach kulek z różnego materiału. Część kulek była wykonana z materiałów jednorodnych (Pb, Al, Si, Ni,Fe, Cu) a część ze stopów 100Cr6, 16MnCr5, CuBe2Co, CuCr, CuCrZr, AlCuMg2. Kulki przed rozpoczęciem właściwego pomiaru docierano, aby usunąć warstwę tlenków. Wpływ twardości badano poprzez zastosowanie kulek hartowanych i niehartowanych wykonanych z materiału 100Cr6, 16MnCr5. Wpływ warstwy tlenków oceniono przez wprowadzenie stalowych kulek hartowanych docieranych i niedocieranych. Kulki smarowane były olejami o różnych lepkościach: n50 = (12, 36, 50, 60, 75, 125 i 200) • mm²/s (cSt). Stwierdzono ubytek materiału z dolnych kulek, częściowo przenoszonego na kulkę górną (rys. 5.62). To spostrzeżenie pozwoliło sformułować równanie objętości strefy aktywnej Va
|
Va = 3 ( Vu + Va + VF )dol + (VAF + VF )górna
| (5.69)
|
Oznaczenia wielkości występujących w powyższym równaniu przedstawiono na rys. 5.62.
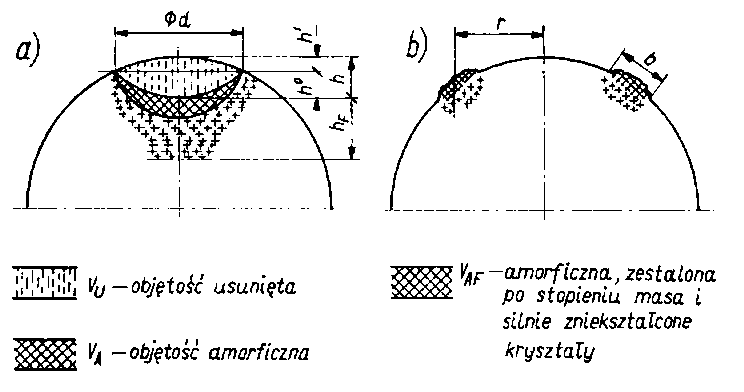 |
Rys. 5.62. Schematyczne przedstawienie objętości zaktywizowanej:
- a) kulki dolne,
- b) kulka górna
|
- Objętość usuniętego materiału Vu można z wystarczającym przybliżeniem określić wagowo lub też obliczyć mając wymiary d i h oraz h` i h (objaśnienia oznaczeń na rys. 5.62)
- Intensywność i głębokość przemian materiału można ocenić ze szlifu metalograficznego i z objętości Vu
|
a = |
Vu + VA + VF
Vu |
; c = |
VAF + VF
Vu
| (5.72)
|
gdzie: współczynniki a i b uzyskuje się z oszacowania głębokości odkształceń dla górnej i dolnych kul. Energię topnienia można obliczyć ze wzoru (5.63) lub też przyjąć z danych w tabl. 5.6.
- Obliczenie pozostałych członów równania (5.67) przedstawia znaczne trudności. Oszacowuje się je na podstawie wartości energii topnienia et. Energię odkształcenia można obliczyć w ten sposób, że pomija się opory tarcia kontaktowego, uwzględnia odkształcenia tylko w jednym kierunku i wówczas można napisać równanie określające zależność pomiędzy siłami zewnętrznymi P a siłami wewnętrznymi
|
h1 | |
r
| |
∫ |
P d h |
= V |
∫ |
kf d r
| (5.73)
| |
h0 | |
0
|
gdzie: ho, h -wymiary ciała, P - siła zewnętrzna, kf -patrz wzór (5.67), V - objętość odkształconego materiału, pozostałe oznaczenia jak poprzednio.
|
Tablica 5.6. Jednostkowa energia topnienia oraz jednostkowa praca zmiany objętości i kształtu (wg Dorna) | |
Materiał | Jednostkowa energia topnienia et
(20°C)
MPa | | | |
Ni | 5780 | 18 | 18 | |
Fe | 5500 | 11 | 18,7 | |
Cu | 3710 | 11 | - | |
Si | 2440 | - | - | |
Al | 1580 | 11 | 23 | | |
Pb | 460 | 11 | 28 | |
CuCr | 3620 | 18 | - | |
CuCrZr | 3620 | 18 | - | |
CuBe2Co | 3350 | 36 | - | |
AlCuMg2 | 1580 | 36 | - | |
16MnCr5 | 6100 | 18 | - | |
16MnCr5 hart. | 6100 | 18 | - | |
10Cr6 hart. | 6100 | 40 do 50 | - | |
ef - jednostkowa energia zmiany kształtu, ev - jednostkowa energia zmiany objętości.
|
- Jednostkowa praca odkształcenia ef jest dla wielu materiałów podana w normie NRD (TGL-28-1001) w zależności od jednostkowego logarytmicznego stopnia odkształcenia r.
- Występuje dobra zgodność (rys. 5.63) pomiędzy wartościami przyjętymi ze wspomnianej normy a wartościami uzyskanymi doświadczalnie. Ze względu na występującą liniową zależność miedzy pracą odkształcenia a energią topnienia jest możliwe wyznaczenie ef z rys. 5.63
|
m ( r ) = f (a1, ... , a4 ) et
| (5.74)
|
gdzie: a1, ... , a3 - pochylenie prostych na rys. 5.63, a4 - pochylenie prostych dla kulek utwardzanych
- Stosunek
|
ef
et |
= (a1, ... , a4 ) r
| (5.75)
|
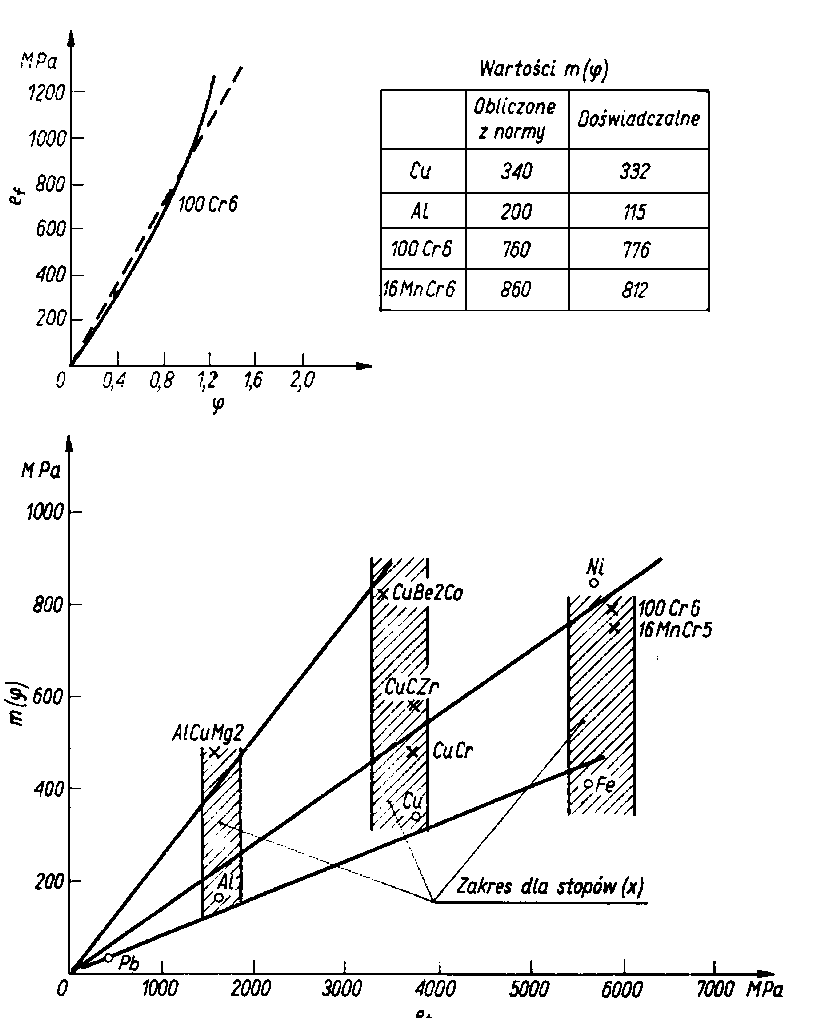 | |
Rys. 5.63. Praca jednostkowa zmiany objętości ef i zależności pomiędzy odkształceniem a energią topnienia et:
a1 » 0,8 (Pb, Al, Cu, Fe),
a2 » 0,13 (Ni, CuCr, CuCrZr, 100Cr6, 16MnCr5),
a3 » 0,26 (AlCuMg², CuBe2Co),
a4 » 0,3 (100Cr6 i 16MnCr5 hartowane)
|
- Należy przyjąć, że około 70% energii odkształcenia przypada na strefę zaktywizowaną Va.
- Energię zmiany objętości można obliczyć z równania (5.64). Znaczną trudność sprawia brak wartości współczynników ściśliwości. Energia zasilania doprowadzona do obszarów tarcia i zużywana na procesy dekohezji oraz zmiany kształtu (odkształcenia) materiału zostaje wg Bowdena i Tabora w 80-94% zamieniona na energię cieplną. Pomiary temperatury wnętrza kuli, wykonane w 0,5 s po jej zatrzymaniu, pozwalają na stwierdzenie, że około 80% ciepła zostaje odprowadzone przez kulę górną i trzpień napędzający. Znaczniejsze odprowadzenie ciepła poprzez olej następuje dopiero wtedy, gdy czas t > tzacier
- Jednostkową energię zasilania można obliczyć z równania (5.62). Można stwierdzić, że dla zakresu obciążenia P < Pzacier Jest ona 3 ¸ 4 razy mniejsza
- Jednostkową energię zasilania można obliczyć z równania (5.62). Można stwierdzić, że dla zakresu obciążenia P < Pzacier Jest ona 3 ¸ 4 razy mniejszaniż dla zakresu obciążeń P > Pzacier. Gęstość energii przypadająca na kule dolne jest wielokrotnie większa niż na kulę górną
|
( ezasil )kul dolnych » ( ezasil )kuli górnej
| (5.76)
|
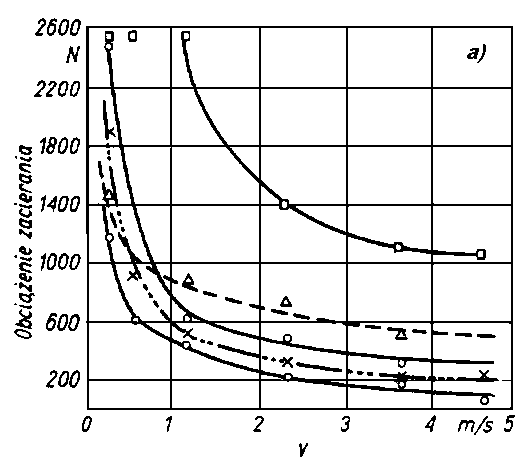
Krytyczna wartość energii jednostkowej powodująca zacieranie jest niezależna od prędkości obrotowej i obciążenia kul. Świadczy to o współzależności prędkości obrotowej i obciążenia. Zależność ta może ulegać zmianom wskutek wpływu takich czynników, jak dokładność obróbki powierzchni, twardość, rodzaj smaru itp. Można ją przedstawić wykreślnie w postaci hiperboli (rys. 5.64) lub zapisać równaniem
gdzie: a, b, c - stałe.
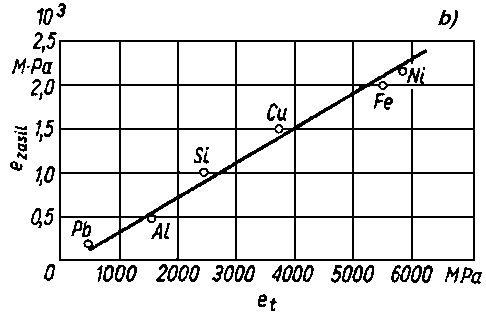 |
Rys. 5.64. Zależność między:
- a) obciążeniem zacierania Pz a prędkością v dla różnych olejów,
- b) energią jednostkową zasilania ezasil przy zacieraniu a energią topnienia materiału kulek et, (wg Dorna)
|
- Jak widać z rys. 5.64 krytyczna jednostkowa energia zasilania przy zacieraniu jest proporcjonalna do jednostkowej energii topnienia et. Odchylenia energii obserwowane w przypadku stopów wynikają głównie z wpływu jaki mają składniki stopowe na wytrzymałość na odkształcenie materiału.
- W omówionych różnych sposobach oceny wytrzymałości warstwy granicznej w warunkach jej niszczenia wykorzystuje się efekty końcowe procesu tarcia, tj. wartość zużycia i obciążenie zacierania. Dla właściwej oceny odporności warstwy oleju na działanie czynników niszczących ważne jest określenie tego, co dzieje się z warstwą oleju w samym procesie tarcia, gdy następuje przerywanie warstwy, jak intensywnie przebiega zużywanie i jaka jest tendencja do regeneracji warstwy.
- Dla uzyskania odpowiedzi na postawione powyżej pytania A. Wachal (100), (101) wprowadził ocenę odporności warstwy oleju opierając się na wykresie momentu tarcia w funkcji czasu. Do pomiaru wykorzystano aparat czterokulowy.
- Wykres momentu tarcia stanowi podstawę oceny trwałości i wytrzymałości warstwy oleju oraz umożliwia ocenę pracy zużywania po przerwaniu warstwy.
Wykres momentu tarcia (rys. 5.65) można podzielić na trzy etapy:
- etap-niszczenia warstwy granicznej,
- etap-zużywanie elementów współpracujących,
- etap - ustabilizowanych oporów tarcia.
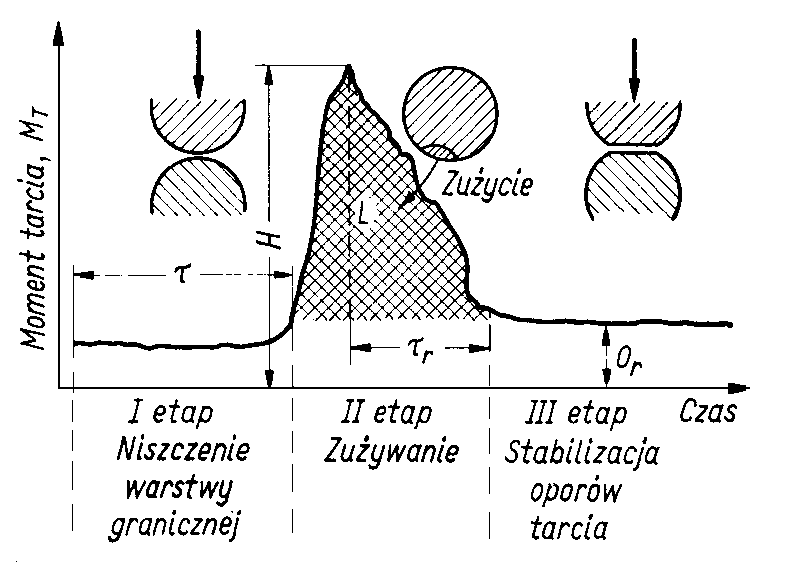 | |
Rys. 5.65. Wykres momentu tarcia w funkcji czasu
|
- Kryteriami oceny pierwszego etapu tj. niszczenia warstwy granicznej, są:
- trwałość warstwy granicznej (czas t od chwili rozpoczęcia pomiaru do początku gwałtownego przyrostu momentu tarcia, świadczącego o przerwaniu warstwy granicznej),
- wytrzymałość warstwy granicznej H (wartość rzędnej momentu w chwili przerywania); ze względu na to, że przerywanie odbywa się na mikropowierzchniach, moment tarcia jest tym większy im na większej liczbie mikronierówności warstwa graniczna jest zniszczona; im wytrzymalsza warstwa graniczna, tym mniejszy moment tarcia w chwili początku przerywania.
- Kryteriami oceny d r u g i e g o e t a p u, tj. zużywania elementów współpracujących, są:
- praca włożona na zużycie L charakteryzowana polem pod krzywą obrysowującą pik momentu,
- zdolność regeneracji warstwy granicznej charakteryzowana czasem powrotu do tarcia w warunkach ustalonych tr i wartością średnią momentu tarcia w czasie etapu zużywania.
- Kryterium oceny t r z e c i e g o e t a p u, tj. ustabilizowanych oporów tarcia, jest wartość momentu tarcia MT. W trzecim etapie powstają warunki do wytworzenia „łożyska ślizgowego” pracującego w zakresie tarcia hydrodynamicznego.
- Wydaje się, że zaproponowany sposób badania odporności warstwy oleju na działanie czynników niszczących daje wiele informacji dotyczących zachowania się warstwy granicznej w czasie tarcia. Duża przydatność tego sposobu do oceny odporności warstwy została wykazana w przeprowadzonych badaniach wpływu dodatków na zachowanie się oleju w warunkach tarcia granicznego (132), jak i wpływu innych substancji tworzących się lub przedostających się do oleju w czasie jego pracy w skojarzeniach trących (133), (134).
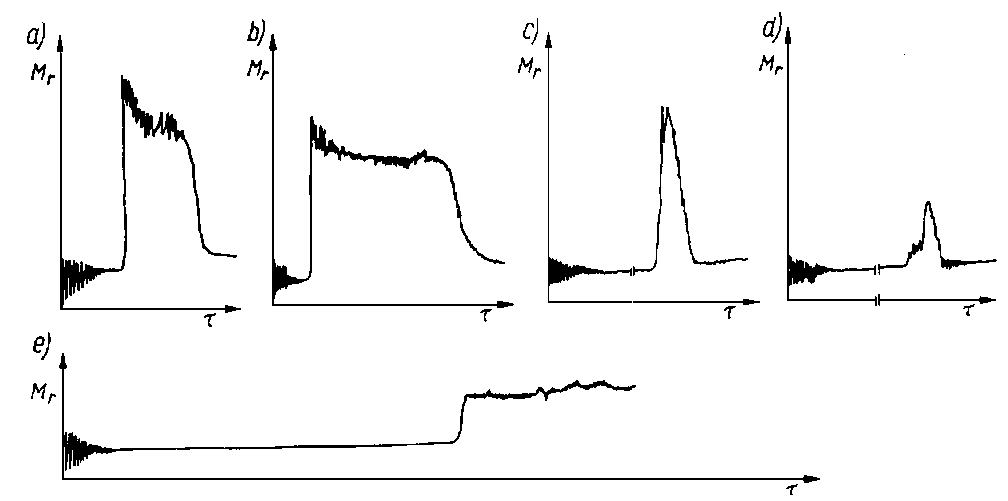 |
Rys. 5.66. Wpływ rodzaju dodatków wprowadzonych do oleju na przebieg krzywych momentu tarcia:
- czysty olej bazowy P-4,
- olej z dodatkiem kwasu palmitynowego,
- olej z dodatkiem chloroparafiny,
- olej z dodatkiem dwualkilo-dwutiofosforanu cynku,
- olej z dodatkiem siarki
|
- Dla przykładu przedstawiono na rys. 5.66 wpływ różnego rodzaju dodatków o różnych momentach dipolowych i różnych zdolnościach sorpcyjnych na wytrzymałość warstwy granicznej, pracę włożoną na zużycie oraz zdolność regeneracji warstwy granicznej.
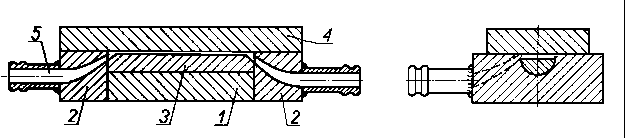
- W celu uniknięcia efektów hydrodynamicznych i efektów wynikających z procesów zużywania, autorzy książki zaproponowali pomiar odporności warstwy granicznej oleju w warunkach statycznych (103). Wstępnym etapem było
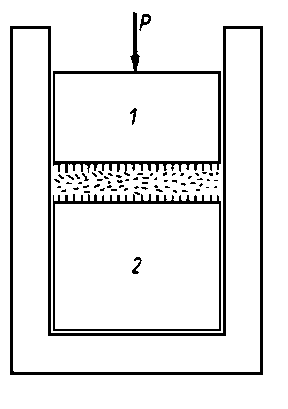 |
Rys. 5.67. Schemat urządzenia do pomiaru wytrzymałości statycznej warstwy oleju;
- 1, 2 - cylindryczne bloki metalowe
|
stwierdzenie, czy w warunkach statycznych istnieją wymierne różnice między wytrzymałością warstwy granicznej oleju czystego i oleju z różnymi dodatkami smarnościowymi. Do tego celu skonstruowano prototyp aparatu (rys. 5.67) składający się z dwóch cylindrycznych elementów metalowych wbudowanych w obejmę z tworzywa sztucznego, spełniającą rolę prowadnicy zapewniającej równoległość powierzchni między blokiem A i B. Między obydwa bloki wprowadzono badany olej i mierzono siłę potrzebną do przerwania warstwy granicznej. Miarą przerwania warstwy była wartość oporu elektrycznego warstwy, która w chwili przerwania musiała być równa oporowi przy styku czystych metalicznych powierzchni.
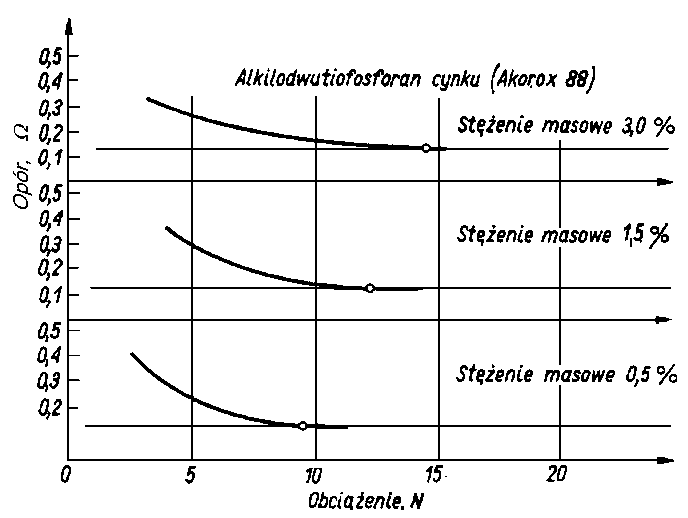 | |
Rys. 5.68. Wytrzymałość statyczna warstwy olejowej przy różnych stężeniach dodatku alkilodwutiofosforanu cynku (Akorox 88)
|
|
Kula | Pierścień | Płytka | |
Kula |  |  |  | |
Pierścień |  | |  | |
Płytka |  |  |  | |
Rys. 5.69. Różne rodzaje skojarzeń tarciowych
|
- Na rys. 5.68 przedstawiono wyniki pomiarów wytrzymałości warstwy granicznej oleju bazowego P-4 przy różnym stężeniu alkilo-dwutiofosforanu cynku (Acorox-88).
- Przy omawianiu metod pomiaru wytrzymałości warstwy oleju należy stwierdzić, że pomiary te niekoniecznie muszą być prowadzone przy kontaktowaniu się elementów stykających się punktowo (np. kul). Wytworzenie warunków tarcia granicznego osiąga się również łatwo przy styku liniowym. Różne rodzaje geometrii styku skojarzeń trących przedstawiono poglądowo na rys. 5.69.
- Podstawowe pojęcia z zakresu smarności i smarowania granicznego zebrano w tabl. 5.7.
|
Tablica 5.7. Określenia związane ze smarnością | |
Polskie | Angielskie | Niemieckie | Rosyjskie | |
Smarność | Oiliness
Lubricity | Schmierfähigkeit
Schmierergebigkeit
Schlüpfrigkeit | Macляиcтcть
Cмaзoчнaя cпocoбнocть
Липкocть | |
Wytrzymałość filmu olejowego | Film strenght | Trägfähigkeit des Schmierfilmes
Filmfestigkeit | Пpoчнocть мacлaнoй плёнки | |
Tarcie graniczne | Boundary friction | Grenzreibung | Гpaничнoe тpeниe | |
Smarowanie graniczne | Boundary lubrication | Grenzschmierung | Гpaничнaя cмaзкa | |
Własności przeciwzużyciowe | Antiwear properties | E. P-Eigenschaften
Verschleiß-verminderung-eigenschaften | Пpoтивизнocныe cвoйcвa | |
Własności przeciwzatarciowe | Antiseizure properties
Antiwelding properties | Freßverminderungeigen-schaften | Прoивизaдныe cвoиcтвa | |
Dodatki smarnościowe | Oiliness improver
Oiliness carriers
E.P. Additives
Antiweld Additives | Hochdruckzusätze
Hochdruckwirkstoffe
E.P. Wirkstoffe | Пpoтивизнocныe и прoивизaдныe пpиcaдки
|
5.3.4. Smarność w ujęciu termodynamicznym
Przyjęcie definicji smarności zaproponowanej przez autorów książki prowadzi do określonego sposobu rozumowania w rozwiązywaniu problemów tarcia granicznego. Jeżeli przyjmie się, że miarą smarności jest wytrzymałość warstwy granicznej na działania niszczące, to możemy znaleźć taką krytyczną wartość oddziaływań niszczących, która powoduje przerwanie warstwy. Wartość tę możemy wyrazić energią lub pracą jednostkową (przypadającą na jednostkę powierzchni). Energia ta powoduje zniszczenie warstwy granicznej, do której wytworzenia układ ma mniejsze lub większe predyspozycje. Predyspozycje te wiążą się ściśle z własnościami podłoża metalowego i własnościami substancji smarnej. Podatność do tworzenia warstwy, a więc do procesów sorpcyjnych, musi być większa lub co najmniej równa energii oddziaływań niszczących. Przyjmując powyższe sformułowania za słuszne M. Dębicki zaadoptował dla procesów tworzenia i niszczenia warstwy granicznej podstawowe prawa termodynamiki chemicznej (105), (106). Uważa on, że wytworzenie warstwy granicznej w wyniku procesów sorpcyjnych może być wyrażone wartością zmiany potencjału termodynamicznego układu
gdzie: G - potencjał termodynamiczny układu, H - entalpia, T - temperatura, S - entropia.
- Ujemna wartość D G daje prawdopodobieństwo samorzutnego przebiegu procesów sorpcyjnych i może być miarą powinowactwa substancji smarnej (lub zawartych w niej dodatków) do podłoża. Procesy chemiczne zazwyczaj są odnoszone do jednostki masy M (zwykle do mola substancji) i rozpatrywane w jakimś określonym przedziale czasu t. Możemy więc zapisać
|
Sorpcja = - |
D G
M t
| (5.79)
|
Ze względu na to, że proces sorpcyjny nie przebiega w rzeczywistości ze 100-proc. wydajnością stosunek sorpcji rzeczywistej do teoretycznie możliwej będzie wartością mniejszą od jedności
|
Sorpcja rzeczywista
Sorpcja teoretyczna |
= C < 1
| (5.80)
|
- W podobny sposób tylko ze znakiem przeciwnym można zapisać procesy desorpcji
|
Desorpcja = + |
D G
M t
| (5.81)
|
- W skojarzeniach trących głównymi czynnikami niszczącymi warstwę zaadsorbowaną i przeciwdziałającymi sorpcji jest obciążenie jednostkowe wyrażone ciężarem na jednostkę masy sorbatu (np. MN/mol) i względna prędkość poślizgu powierzchni trących (np. w metrach na czas t). Jeżeli tworząca się sorbowana warstwa graniczna ma być trwała, to potencjał termodynamiczny tworzenia ( D G/ M t ) musi być co najmniej równy oddziaływaniom niszczącym, a więc iloczynowi obciążenia i prędkości poślizgu P v . Iloczyn ten oznacza pracę w czasie (czyli moc) przypadającą na jednostkę powierzchni warstewki smarnej między ciałami trącymi i narzucanej układowi trybologicznemu z zewnątrz
|
P v = |
D G
M t |
; ( P, T = const )
| (5.82)
|
gdzie t - czas sorpcji (tworzenia warstwy granicznej).
- Jak widać z powyższego równania smarność można wyrażać w przypadku tworzenia warstwy granicznej prawą stroną równania, a w przypadku jego niszczenia lewą stroną równania. Jeżeli D G wyrazimy w kJ, to smarność możemy określić wartością efektu cieplnego przypadającego na jednostkę masy sorbującej substancji w jednostce czasu, a więc w kJ/(mol•s). Przy niszczeniu warstwy granicznej smarność możemy wyrazić mocą jednostkową (przypadającą na jednostkę powierzchni) dostarczoną do układu trybologicznego z zewnątrz.
- Opierając się na obydwu sformułowaniach można wyprowadzić jednostki smarności.
- Na podstawie wartości potencjału termodynamicznego próbuje Dębicki wytłumaczyć powstawanie płaszczyzn poślizgu. Uważa on, że jeśli warstwa sorbatu utworzyła się wskutek zmiany potencjału -D G, to od strony niezwiązanej z podłożem warstwa ta będzie miała dodatnią wartość potencjału, a więc zetknięcie warstw granicznych doprowadzi do odpychania i do łatwego poślizgu pomiędzy warstwami. A więc łatwy poślizg pomiędzy warstwami powstaje wówczas, gdy D G > 0, czyli
- Opierając się na wartości potencjału termodynamicznego przewiduje również Dębicki możliwość przyspieszania zużywania współpracujących elementów przez wprowadzenie dodatków smarnościowych.
- Jeżeli energia tworzenia warstewki sorpcyjnej zrówna się z energią tworzenia sieci Us wówczas może nastąpić odrywanie atomów powierzchniowych materiału podłoża przez cząsteczki warstwy granicznej
W takim przypadku dodatek smarnościowy stanie się stymulatorem zużywania powierzchni trących. Z powyższego względu energia wiązania cząsteczki sorbowanej nie może przekraczać wartości energii wiązania atomów z siecią macierzystą, chyba żeby usunięty z powierzchni związek ułatwiał poślizg między współpracującymi elementami.
5.3.5. Zależność między budową chemiczną substancji smarujących a ich zdolnością sorpcyjną
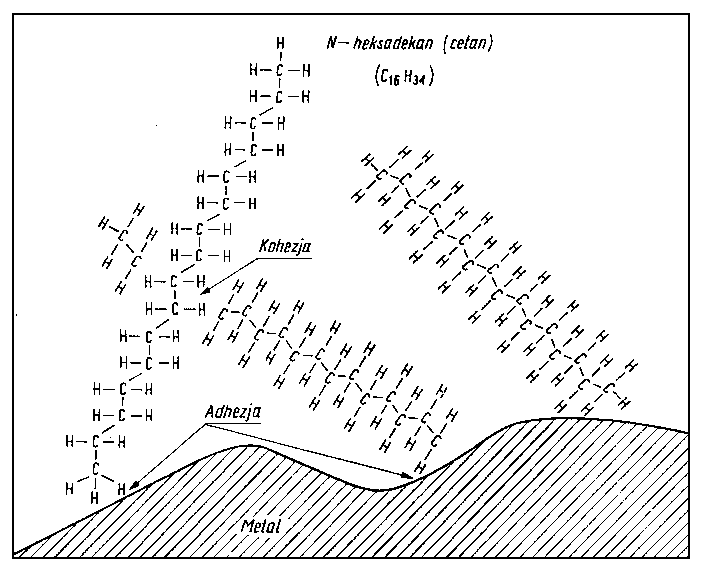
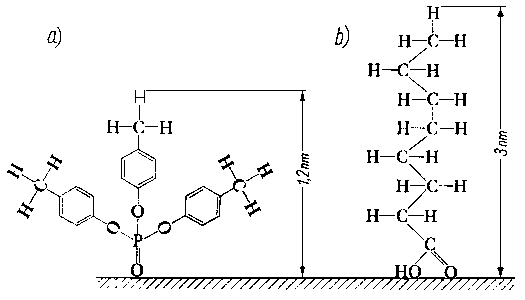
Po omówieniu ogólnych praw adsorpcji i chemisorpcji, zasad tworzenia warstwy granicznej oraz sposobów pomiaru trwałości tej warstwy należy zastanowić się, jak powierzchnie ciał stałych, a zwłaszcza metali, które są podstawowym tworzywem w budowie maszyn, oddziaływują na poszczególne grupy substancji smarujących i jakie istnieją możliwości tworzenia trwałych, silnie związanych z podłożem warstw smarnych.
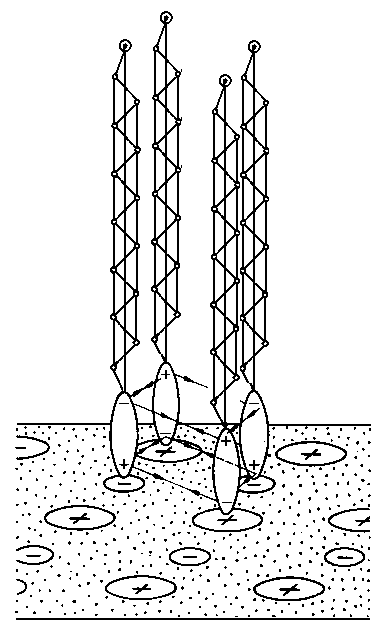
- Nad powierzchnią metalu istnieje pole sił, którego charakter został już uprzednio omówiony. Oddziaływanie pola elektrostatycznego i fali elektrodynamicznej powoduje u cząsteczek zbliżających się do powierzchni zaburzenia w rozkładzie ich ładunków elektrycznych, a więc powstawanie indukowanych momentów dipolowych o wartości zależnej od budowy cząsteczki. Oczywiście intensywniejsze oddziaływanie następuje wtedy, gdy cząsteczka ma trwały moment dipolowy. We wszystkich przypadkach w grę wchodzą oddziaływania siłami dyspersyjnymi (Londona).
- Największy udział tonażowy w światowej technice smarowniczej mają oleje węglowodorowe o specyficznej budowie, na którą składają się elementy struktur aromatycznych naftenowych i parafinowych. W celu omówienia współdziałania z powierzchnią takich złożonych struktur mieszanych należy najpierw omówić, jak takie współdziałanie jest możliwe w przypadku struktur prostych.
- Należy zaznaczyć, że ogólnie węglowodory niezależnie od ich przynależności do poszczególnych rodzin węglowodorowych nie mają zazwyczaj trwałego momentu dipolowego, a jeżeli ze względu na ich budowę przestrzenną moment taki istnieje, to ma on bardzo niewielką wartość. Tak więc w przypadku węglowodorów oddziaływanie siłami orientacyjnymi (Keesoma) albo nie będzie występowało, albo będzie odgrywało bardzo nieznaczną rolę. Większe znaczenie mają w tym przypadku siły indukcyjne (Debye'a) ze względu na powstawanie, w wyniku oddziaływania pola powierzchni metalu, indukowanych momentów dipolowych.
- Przechodząc do węglowodorów parafinowych, a więc węglowodorów o budowie łańcuchowej, należy sobie zdawać sprawę z tego, że oprócz sił dyspersyjnych (Londona) u cząsteczek łańcuchowych mogą występować chwilowe momenty dipolowe w wyniku rotacji poszczególnych członów łańcuchów wokół wiązań pojedynczych. Rotacja ta, w zależności od chwilowego położenia poszczególnych członów łańcucha względem siebie, wywołuje chwilowy nierównomierny rozkład ładunków, a więc i powstawanie chwilowych momentów dipolowych. Rotacja ta może być hamowana różnego rodzaju zawadami przestrzennymi, jak np. rozgałęzieniami łańcucha. Położenie w czasie obrotu poszczególnych członów cząsteczki względem siebie różni się energią potencjalną.
- Zmiany ułożenia przestrzennego atomów w cząsteczce w wyniku rotacji dookoła wiązania pojedynczego są zwane konformacją, natomiast izomery rotacyjne zwane są konformerami.
- Niesymetrycznego rozkładu ładunków w wyniku konformacji można się również doszukiwać i w przypadku węglowodorów naftenowych. Niesymetryczny rozkład ładunków elektrycznych jest tutaj możliwy wskutek niesymetrycznego rozkładu podstawników względem pierścieni, jak również i różnorakich możliwych form deformacji samych pierścieni.
- Dla węglowodorów aromatycznych istnieje możliwość konformacji w przypadku układów kilkupierścieniowych, gdy poszczególne pierścienie są łączone między sobą wiązaniami pojedynczymi (jak np. w dwufenylu). Występowanie elektronów p w pierścieniach aromatycznych stwarza dodatkowe możliwości oddziaływania węglowodorów aromatycznych z powierzchnią metali.
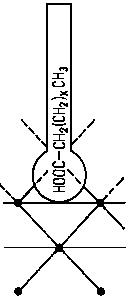
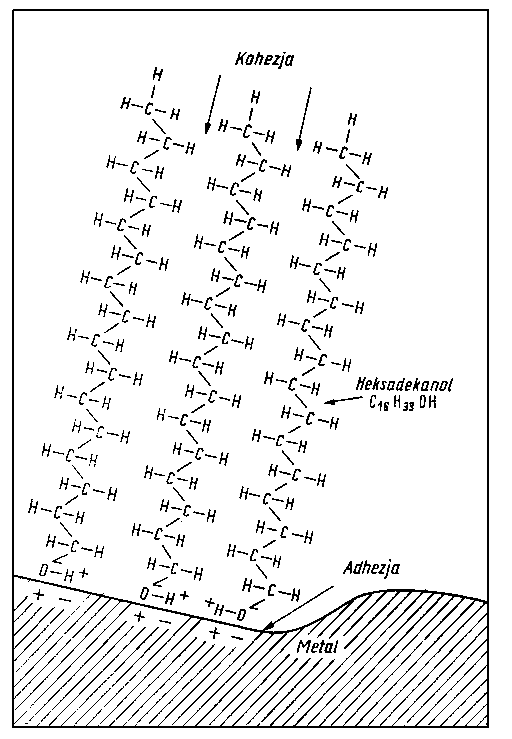
- Dla węglowodorowych struktur złożonych są możliwe wszystkie wspomniane oddziaływania, a więc konformacja łańcuchów węglowodorowych doczepionych do pierścieni, konformacja pierścieni naftenowych wchodzących w skład budowy cząsteczki oraz oddziaływanie elektronów p z polem sił lub atomami powierzchniowymi. Utlenianie się węglowodorów wchodzących w skład oleju, podczas pracy oleju w urządzeniach, powoduje powstawanie związków tlenowych mających mniejszy lub większy moment dipolowy. Związki te jako trwałe dipole mogą swoim polem indukować w niepolarnych cząsteczkach węglowodorów momenty dipolowe. Tak więc oleje po pewnym okresie pracy w urządzeniu wykazują większą skłonność do wiązania się z powierzchnią niż oleje świeże. Należy zaznaczyć, że nawet oleje świeże, jeżeli ich stopień rafinacji nie jest zbyt głęboki, zawierają pewną bardzo nieznaczną ilość „naturalnych" związków polarnych. Należą do nich związki asfaltowo-żywiczne oraz niektóre związki siarkowe i tlenowe. Związki te ułatwiają tworzenie na powierzchniach metali adsorbowanych, zorientowanych warstw cząsteczek węglowodorów.
- Oleje syntetyczne typu dwuestrowych mają silne trwałe momenty dipolowe i dlatego ich trwała adsorpcja na powierzchni jest zjawiskiem oczywistym. Trwałymi momentami dipolowymi obdarzone są również i pozostałe oleje syntetyczne. Spośród nich najmniejszą zdolność oddziaływania na powierzchnię metali mają oleje metylosilikonowe. Skłonność do tworzenia przez nie trwałych adsorbowanych warstw granicznych zwiększa się przez wprowadzanie do tych olejów dodatków o budowie polarnej. Wady tej nie mają oleje fenylosilikonowe.
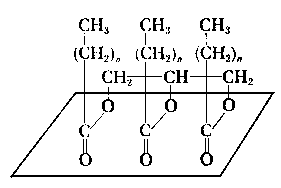
- Adsorpcja i chemisorpcja smarów plastycznych na powierzchni ciał stałych jest zagadnieniem bardzo mało poznanym. Ze względu na to, że smary plastyczne są koloidalnymi roztworami mydeł w olejach węglowodorowych, należy przypuszczać, że mydła kwasów tłuszczowych, jako substancje polarne rozproszone w niepolarnym oleju, powodują uporządkowanie warstwy granicznej. Nie jasny jest jednak wpływ środowiska o bardzo dużej lepkości na swobodę ruchów cząsteczek polarnych ku powierzchni, a ponadto sam mechanizm adsorpcji, nie wiadomo bowiem czy adsorpcji podlegają poszczególne cząsteczki, czy też całe micele tego koloidalnego roztworu.
- Niezbyt jasny jest również mechanizm współdziałania smarów stałych z powierzchnią metali. Dla smarów o budowie płytkowej jest możliwe oddziaływanie typu powierzchnia ciała stałego-powierzchnia ciała stałego przy oparciu się na rozmieszczeniu ładunków powierzchniowych oraz na oddziaływaniu dwóch pól sił. Dla grafitu niektórzy autorzy przyjmują, że jego atomy powierzchniowe, a zwłaszcza atomy naroży i krawędzi jako bogatsze energetycznie, mogą podlegać utlenianiu, co może powodować zwiększenie sił oddziaływania międzypowierzchniowego. Podobną rolę przypisuje się zaadsorbowanej na powierzchniach płytek parze wodnej. Dla dwusiarczku molibdenu również rozważa się wpływ utlenienia powierzchniowego płytek. Niektórzy autorzy uważają, że przy wysokich naciskach występujących w procesie tarcia wyzwala się duża ilość ciepła, która powoduje miejscowe skoki temperatury prowadzące do reakcji chemicznej między atomami siarki płytek dwusiarczku molibdenu a powierzchnią metali. W tej reakcji chemicznej upatrują oni charakter związania dwusiarczku molibdenu z podłożem metalicznym. Wydaje się jednak słuszne traktowanie powyżej przytoczonych sądów, jako niezbyt należycie udokumentowanych doświadczalnie.
5.3.6. Dodatki zwiększające smarność
Oleje mineralne, a więc oleje węglowodorowe, ze względu na swoją obojętność chemiczną oraz brak momentu dipolowego nie są substancjami mogącymi zapewnić wytworzenie trwałej warstwy granicznej. Z tego względu, aby przystosować olej do pracy w warunkach smarowania granicznego, konieczne jest wprowadzenie dodatków smarnościowych, tj. dodatków zwiększających smarność olejów. Muszą one mieć przynajmniej duży moment dipolowy, gdyż wtedy jako trwałe dipole mogą w niepolarnych cząsteczkach węglowodorów oleju wywoływać indukowane momenty dipolowe, co zwiększa ich możliwość oddziaływania z powierzchnią metalu. Energia tego rodzaju oddziaływania ma raczej niewielką wartość rzędu kilkunastu kJ/mol. Jeżeli dodatki smarnościowe są reaktywne chemicznie i mogą tworzyć na podłożu metalu warstwę chemisorbowaną, wówczas energia związania z podłożem jest rzędu około 627 kJ/mol (150 kcal/mol), a więc powstała warstwa jest bardzo trwała.
- Jeżeli dodatki smarnościowe mają ponadto charakter związków powierzchniowo-czynnych, to adsorbują się one na powierzchni oleju tak, że nawet przy bardzo małej ich ilości w oleju zawsze na powierzchni rozdziału występują one w dużym stężeniu i wskutek tego przy kontakcie oleju z powierzchnią metalu mogą z nią reagować.
- Większość dodatków smarnościowych stosowanych obecnie ma zdolność do chemisorpcji. W literaturze patentowej można znaleźć tysiące związków sugerowanych jako dodatki smarnościowe. Tylko nieznaczna część z nich znalazła praktyczne zastosowanie. Dodatki smarnościowe są zazwyczaj związkami siarkowymi, chlorowymi, fosforowymi lub tlenowymi.
5.3.6.1. Związki tlenowe
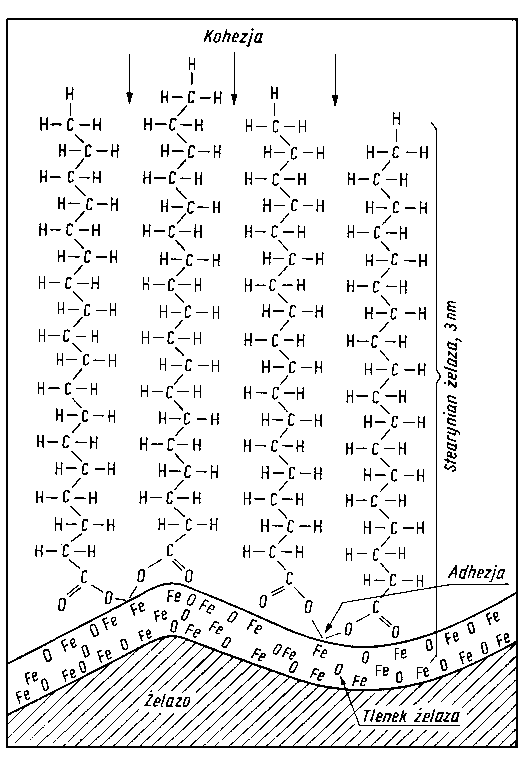
Najstarszymi dodatkami smarnościowymi są tłuszcze (czyli glicerydy kwasów tłuszczowych). Dawno temu wprowadzono stosowanie olejów natłuszczanych. Oleje tego typu stosuje się do dziś do smarowania niektórych urządzeń. W okresie międzywojennym jako olej lotniczy o dużej smarności stosowany był olej rycynowy, a więc tłuszcz roślinny. Obecnie natłuszczanie olejów stosuje się rzadko z tego względu, że tłuszcze podczas pracy oleju w urządzeniach mogą wydzielać substancje kwaśne, które oddziaływują korodujące na metal. Z powyższych przyczyn bardzo ograniczone jest stosowanie kwasów tłuszczowych jako dodatków. Trochę większe znaczenie mają ich estry w tych przypadkach, gdy nie zachodzi obawa ich zmydlenia i wydzielenia wolnego kwasu. Oddziaływania korozyjnego nie mogą wykazywać sole kwasów tłuszczowych, toteż stosowane są one jako dodatki smarnościowe, tzw. łagodne (mild), wytrzymałe na wysokie naciski jednostkowe. Największe zastosowanie mają sole ołowiowe kwasów tłuszczowych, jak np. oleinian ołowiu, stearynian ołowiu i natleniany ołowiu (stosowane jako dodatki smarnościowe do olejów przekładniowych).
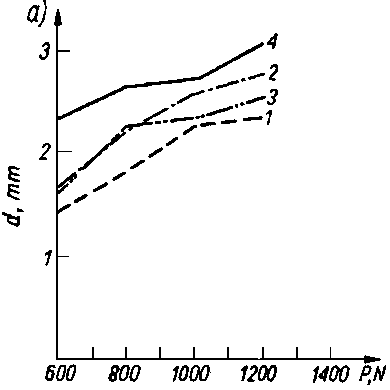
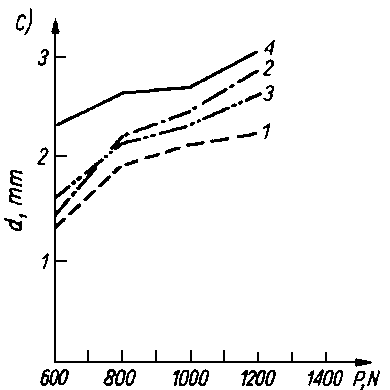
- Kwasy tłuszczowe jak i długo łańcuchowe alkohole były przedmiotem obszernych badań nad trwałością warstwy granicznej. Zwiększają one bardzo nieznacznie własności przeciwzatarciowe oleju, natomiast obniżają zużycie oraz opory tarcia (rys. 5.70).
5.3.6.2. Związki siarkowe
Osobną grupę dodatków smarnościowych stanowią związki siarkowe. Czynnikiem warunkującym aktywność tych związków jest siarka jako element ich budowy. Związki te mogą być albo indywidualnymi związkami siarkowymi, albo też mogą to być siarkowane mieszaniny. Istnieje również możliwość wprowadzania siarki elementarnej do oleju. Po ogrzaniu oleju pewna jej ilość (do około 2%) rozpuszcza się w oleju. Powstają w ten sposób tzw. oleje siarkowane. Dotychczas nie zbadano czy siarka jedynie rozpuszcza się w oleju, czy też wchodzi w reakcję chemiczną ze składnikami oleju. Oleje siarkowane są stosowane przy obróbce metali skrawaniem jako ciecz smarująca, jak również dla przyspieszenia docierania urządzeń i maszyn.
a ) alkohol decylowy, | 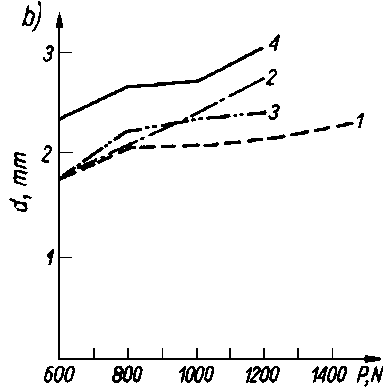
b ) alkohol cetylowy, |
c ) alkohol oktadecylowy, | 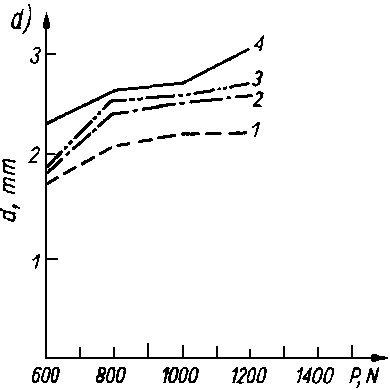
d ) kwas laurynowy, |
e ) kwas palmitynowy, | 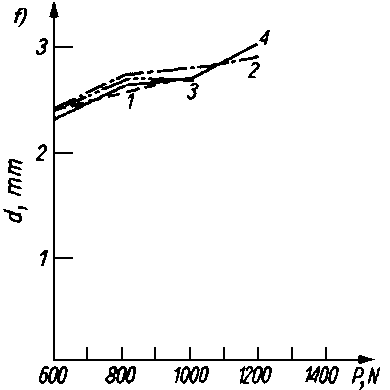
f ) kwas stearynowy, |
g ) kwas rycynowy, | 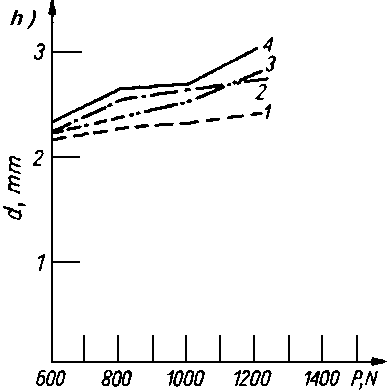
h ) kwas oleinowy; |
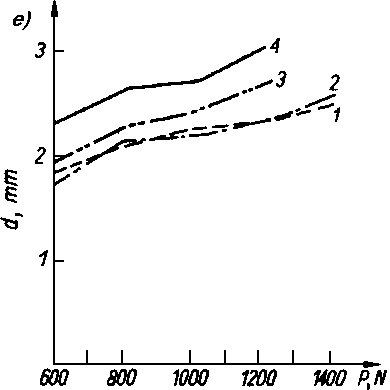
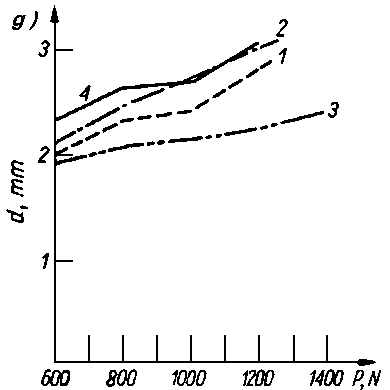
Rys. 5.70. Odporność warstwy olejowej oleju bazowego P-4 czystego i z dodatkami alkoholi i kwasów tłuszczowych mierzona za pomocą aparatu czterokulowego:
- d — średnica śladu zużycia,
- P — siła obciążenia.
|
stężenie dodatku
- — 0,5%,
- — 1,5%,
- — 3,0%,
- — czysty olej P-4,
|
- Siarkowane mieszaniny
W wyniku działania siarki lub jej związków (np. chlorku siarki) na mieszaniny związków organicznych powstają siarkowane mieszaniny, często efektywnie stosowane jako dodatki smarnościowe. Od dawna stosowane są siarkowane tłuszcze, a zwłaszcza siarkowane oleje roślinne, jak np. siarkowany olej rzepakowy, olej konopny, olej palmowy itp. Oleje tłuszczowe zawierające estry nienasyconych kwasów tłuszczowych stosunkowo łatwo wiążą siarkę do wiązań nienasyconych. Siarka tworzy w tych związkach mostki siarkowe
O
║
CH3(CH2)CH═CH—CH—(CH2)x—C—OR
│
S2 O
│ ║
CH3(CH2)CH═CH—CH—(CH2)x—C—OR
|
lub
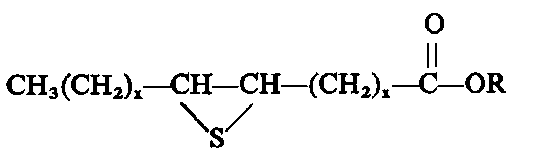
|
- Oprócz tłuszczy siarkowanych, tj. naturalnych estrów (glicerydów) nasyconych i nienasyconych kwasów tłuszczowych, otrzymuje się również estry metylowe, etylowe i wyższe tych kwasów, które następnie poddaje się siarkowaniu. Przykładem może być krajowy dodatek SEKT (siarkowany ester kwasów tłuszczowych).
- Oddzielną grupę siarkowanych mieszanin stanowią siarkowane polimery. Przykładem może tu być siarkowany poliizobutylen występujący pod handlową nazwą Anglomol 32, Elco 217. Występuje on również jako składnik Anglomolu 99 używanego jako dodatek do olejów przekładniowych. Stosowane są również siarkowane tetramery propylenu.
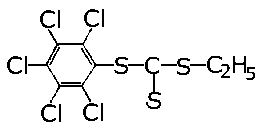
- W Związku Radzieckim otrzymuje się w wyniku działania siarki elementarnej na mieszaninę polimerów węglowodorów olefinowych C3 - C5 dodatek noszący handlową nazwę OTP. Jest to ciecz o kolorze ciemnobrązowym o masie cząsteczkowej około 200, składająca się głównie (ok. 65%) z dwualkilodwusiarczków oraz siarczków alkilowych (rys. 5.71).
- W szerokim zakresie są również stosowane siarkowane terpeny jako dodatki do olejów przekładniowych, jak również do olejów silnikowych. Oprócz
 |
Rys. 5.71. Zależność zużycia od obciążenia dla olejów z dodatkami siarkowymi;
- - siarkowany olej przekładniowy,
- - olej PS-28 czysty,
- - PS-28+5% siarkowanego oleju konopnego,
- - olej TS-14,5, czysty,
- - TS-14,5+mieszanina jedno- i dwusiarczków alkilowych,
- - TS-14,5+5% dwusiarczku dwufenylu,
- - TS-14,5+10% siarkowanych estrów kwasów tłuszczowych,
|
- - TS-14,5+10% siarkowanego oktolu,
- - TS-14,5+5% dwusiarczku dwubenzylu,
- - TS-14,5+3% dwusiarczku dwualkilobenzylu,
- - TS-14,5+3% dwualkilo-benzylotioetanu,
- - TS-14,5 + 3% siarkowanych olefinów (dodatek OTP),
- - TS-14,5 + 3% siarkowanego dwusiarczku dwubenzylu (dodatek OD-3),
- - TS-14,5+3% siarkowanych terpenów, 7.5-TS-14,5+3% dwusiarczku dwubenzylu
|
działania smarnościowego uważa się, że mają one również działanie przeciwkorozyjne.
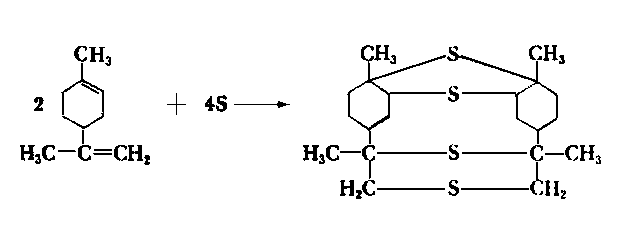
- W kraju w latach sześćdziesiątych w handlu znajdowały się silnikowe oleje Extra ST i Extra 8T zawierające krajowy dodatek terpenowy. Jego produkcja miała wiele niedociągnięć i dodatek ten łatwo wydzielał się z oleju, a ponadto łatwo ulegał zeżywiczeniu, co prowadziło do zapiekania pierścieni tłokowych. Jak podaje Winogradowa (44) w Związku Radzieckim siarkowane terpeny są składnikiem wieloskładnikowego dodatku przeciwzatarciowego EZ-5. Stwierdzono, że dodatek ten w niewielkim stopniu działa jako dodatek przeciwzużyciowy, natomiast dobrze jako dodatek przeciwzatarciowy. Lepsze efekty uzyskuje się, gdy zamiast działania siarką elementarną na terpeny, używa się chlorku siarki. Siarkowane w ten sposób terpeny zawierają 15- 17% mas. siarki oraz 3-4% mas. chloru, co nadaje im bardzo dobre przeciwzatarciowe własności. Mają one handlową nazwę OT-1 (osiernienyje tierpeny).
- Według Winogradowej (44) w wyniku działania siarki na terpeny tworzą się następujące związki:
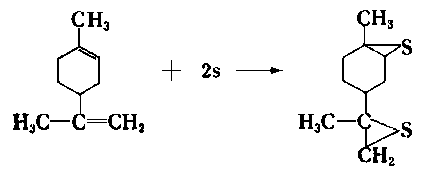
|
|
- Siarczki i dwusiarczki
Siarczki i dwusiarczki będące organicznymi związkami siarki znalazły zastosowanie jako dodatki smarnościowe. Ogólnie budowę tych związków można przedstawić następująco:
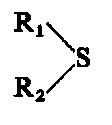 siarczki |
R1—S—S—R2 dwusiarczki
|
- Jako R1 i R2 mogą występować różnego rodzaju podstawniki alkilowe, cykloalkiIowe i arylowe. Spośród dwusiarczków najczęściej stosowane są:
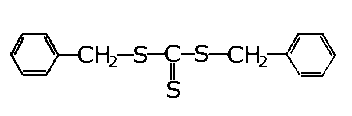
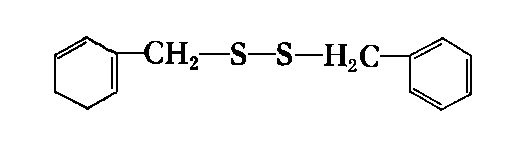 dwusiarczek dwubenzylu | |
|
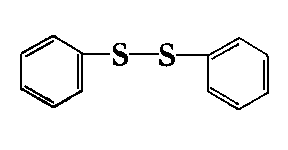 dwusiarczek dwufenylu | |
|
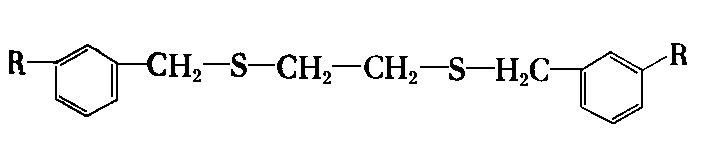 dwualkilotiobenzylo etan | |
|
- Najlepszymi własnościami odznacza się dwusiarczek dwubenzylu. Były również robione próby zastosowania dwusiarczku dwuheksylu, który okazał się efektywny. W Związku Radzieckim dla zwiększenia zawartości siarki w dwusiarczku dwubenzylu, traktowano go dodatkowo siarką w podwyższonej temperaturze. Pozwala to na uzyskanie trój- i czterosiarczków benzylowych. Mieszanina tych związków, jako dodatek noszący nazwę OD-3, jest stosowana w procesach docierania. Allum z współpracownikami (107), (108) badał własności smarnościowe dwusiarczków organicznych. Badaniu podlegały dwusiarczki: dwuetylowe, n-dwubutylowe, n-dwuoktylowe, n-dwudodecylowe, n-dwuheksadecylowe i dwu n-dwuoktadecylowe. Stwierdzono zwiększenie się własności przeciwzużyciowych wraz ze wzrostem długości łańcucha podstawnika alkilowego. W badaniach wpływu rodzaju podstawnika stwierdzono następujące uszeregowanie podstawników: fenylowy < butylowy < sec-butylowy < tert-butylowy < benzylowy.
- Oprócz wymienionych na wstępie dwusiarczków z podstawnikami arylowymi, inne dwusiarczki nie znalazły w szerszym zakresie zastosowania jako dodatki smarnościowe. Stwierdzono, że dodatki typu dwusiarczków działają efektywniej w obecności innych dodatków zawierających chlor (synergia).
- Ksantogeniany i tiowęglany
Ksentogeniany i tiowęglany są pochodnymi następujących związków:
HO—C—SH
║
S
kwas dwutiowęglowy |
RO—C—SH
║
S
kwas ksantogenowy |
RO—C—SR
║
S
ksantogenian
|
Po raz pierwszy ksantogeniany jako dodatki smarnościowe zastosowali Niemcy w okresie drugiej wojny światowej pod nazwą Mesulfolu I i Mesulfolu II. Pierwszy był produktem reakcji ksantogenianu sodu z alkoholem izoamylowym, drugi z dwuchloroetanem. Obecnie stosowanych jest szereg tego typu związków. Tak np. w Związku Radzieckim opracowano serie dodatków ksantogen łanowych noszących różne handlowe nazwy. Mają one dobre własności przeciwzatarciowe, a ponadto działają antyutleniająco (rys. 5.72).
 |
Rys. 5.72.Zależność zużycia od obciążenia dla oleju z dodatkami estrów tiowęglowego (we Winogradowej);
- - olej TS-14,5 czysty,
- - +5% dodatku LZ 6/9,
- - +5% dodatku LZ-23K,
- - +5% ksantogenianu dwubutylu,
- - +5% trójtiowęglanu dwubutylu,
- - +10% dodatku LZ-20,
- - +10% dwuksantogenianu etylenglikolu
|
(izo-C5H11O—C—S—CH2)2
║
S
- Dodatek LZ-19
|
(izo-C4H9O—C—S—CH2)2
║
S
- Dodatek LZ-25K
|
(n-C4H9O—C—S—CH2)2
║
S
- Dodatek LZ 6/9
|
(izo-C3H7OC—S—CH2)2
║
S
- Dodatek LZ-23K
|
(C2H2OC—S—CH2)2
║
S
- Dodatek LZ-24
|
Dodatkowe działanie antyutleniające zwiększa się w przypadku podstawników fenylowych.
- Osobną grupę stanowią trójtiowęglany organiczne bardzo rozpowszechnione w krajach zachodnich, dobrze rozpuszczalne w olejach, przejawiające ponadto działanie przeciwutleniające i przeciwkorozyjne. W przypadku ograniczonej rozpuszczalności związku w oleju, zwiększenie rozpuszczalności osiąga się poprzez wprowadzenie atomów chloru do cząsteczki. Poniżej przedstawiono kilka przykładów trójtiowęglanów
- Dwutiokarbaminiany
Tego typu dodatki w swojej cząsteczce oprócz atomów siarki mają również azot. Dużą serię dwutiokarbaminianów metali, jako dodatków do olejów, wypuściła na rynek amerykańska firma Vanderbilt Co. pod nazwą Vanlube. Poniżej podano kilka przykładów tego typu dodatków
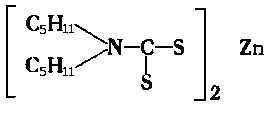 Dodatek Vanlube AZ |
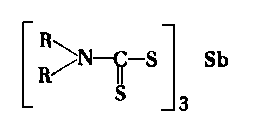 Dodatek Vanlube 73
|
(sól kadmu tego samego związku ma nazwę Vanlube 61, a sól ołowiowa-Vanlube 71).
- Inne związki siarkowe
Merkaptany i tiokwasy, pomimo że ze względu na swoją reaktywność podnoszą odporność warstwy olejowej, nie są praktycznie stosowane, ponieważ pogarszają odporność oleju na utlenianie, a ponadto oddziaływują korozyjnie zwłaszcza na metale nieżelazne. Dobrymi własnościami smarnościowymi odznaczają się heterocykliczne związki siarkowe, otrzymane w wyniku oddziaływania siarki na olefiny w temperaturze 160-300°C. Najogólniej ich budowę można przedstawić wzorem
 |
(R, R1 = H, alkil lub aryl)
|
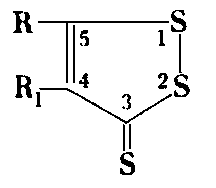
Mają one potoczną nazwę ditioltiony. Przykładem takiego związku może być 4-neopentylo-5-tert-butylo-l,2-dwutiocyklopenten-4-tion-3 (zwany dodatkiem NPT)
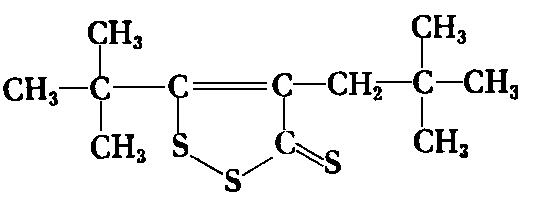
|
- Ditioltiony z podstawnikami aromatycznymi są trudno rozpuszczalne w olejach, ale można je wprowadzać w roztworze w specjalnych rozpuszczalnikach, natomiast ditioltiony mające podstawniki alifatyczne oraz otrzymane z terpenów są dobrze rozpuszczalne i mają dobre własności smarnościowe (rys. 5.73).
 |
Rys. 5.73.Zależność zużycia od obciążenia dla oleju z dodatkami dwutioltionów (wg Winogradowej);
- - olej TS-14,5 +3% 4-neopentylo 5-IlI-rz. butylo-l,2-dwucyklopenteno-4-tionu-3;
- - olej TS-14,5 i dodatek LZ 36/1 w stosunku 1:1 +2,5% mas. 4,5-dwufenylo-l,2-dwucyklopenteno-4-tionu-3;
- - olej PS-28 + 3% mas. ditioltionów otrzymanych z terpenów
|
 |
Rys. 5.74. Zależność zużycia od przyłożonego obciążenia (wg Kulczyckiego);
- - hydrorafinat-2,
- - hydrorafinat-2 + merkaptan cetylowy,
- - hydrorafinat-2 +b-tionaftol,
- - hydrorafinat-2 +dwusiarczek dwufenylu,
- - hydrorafinat-2 + tiobenzofenon,
- - hydrorafinat-2 + dwusiarczek dwubenzylu,
- - hydrorafinat-2 + siarka elementarna (stężenie dodatków w przeliczeniu na 0,5% mas. siarki elementarnej)
|
- Były również robione próby zastosowania jako dodatków smarnościowych innych związków siarkowych, tioketonów, triobenzofenonu, tionaftolu, pochodnych tiofenu i wiele innych związków (rys. 5.74), ale jak dotychczas nie znalazły one zastosowania w szerszym zakresie.
5.3.6.3. Związki chlorowe
Związki chlorowe jako dodatki smarnościowe tworzą na powierzchniach metali trwałe i odporne na przerwanie warstwy chemisorbowane. Przy stosowaniu większości związków chlorowych istnieje niestety obawa, że produkty ich rozkładu mogą stać się przyczyną korozji smarowanego metalu. Zwłaszcza niebezpieczne są te związki które mogą odszczepiać chlorowodór.
- Najbardziej rozpowszechnionym chlorowym dodatkiem smarnościowym jest chloroparafina. Otrzymuje się ją przy przepuszczaniu gazowego chloru przez stopioną parafinę. Handlowa chloroparafina zawiera około 40% mas. przyłączonego chloru. Znacznie mniejsze zastosowanie znalazły inne chlorowane produkty naftowe, jak chlorowana nafta i chlorowane oleje, natomiast stosunkowo często są stosowane chlorowane mieszaniny różnych związków organicznych. Tak np. chlorowaniu poddaje się terpeny (gorsze gatunki kalafonii), różne rodzaje wosków i żywic, tłuszcze, nienasycone kwasy tłuszczowe, estry, alkohole o długich cząsteczkach oraz niektóre polimery o niskim stopniu spolimeryzowania. Dokonywano prób zastosowania niskopolimeryzowanego polichlorku winylu oraz sześciochlorobutadienu. W jednym z patentów proponuje się użycie estrów kwasu czterochloroadypinowego, czterochlorosebacynowego i pięciochloroolejowego. Spośród chlorowych pochodnych węglowodorów bardzo dobre efekty uzyskuje się przy chloroalkanach i chlorocykloalkanach, a znacznie gorsze przy chlorowanych węglowodorach aromatycznych. Przykładem stosowanych, jako dodatki, chlorowanych węglowodorów może być sześciochloroetan Cl3C—CCl3, sześciochlorocykloheksan, sześciochlorocyklopentadien i ośmiochlorocyklopenten.
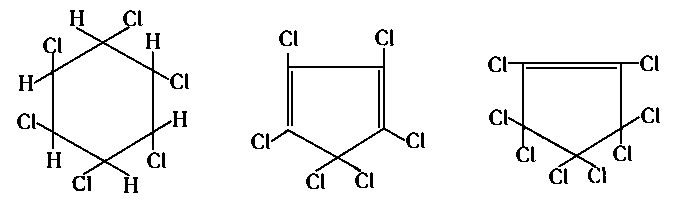
|
- W Związku Radzieckim pod nazwą Sowol stosuje się mieszaninę cztero- i pięciochloro podstawionych pochodnych dwufenylu. Przy zastosowaniu Sowolu uzyskuje się nieco gorsze efekty niż w przypadku chloroparafiny (rys. 5.75).
5.3.6.4. Związki fosforowe
Za bardzo efektywne, a nie powodujące oddziaływania korozyjnego, uważane są związki fosforowe. Najczęściej stosowane są różnego rodzaju alkilowe pochodne kwasu fosforowego i fosforawego. Alkilofosforyny są uważane za bardziej efektywne
 |
Rys. 5.75. Zależność zużycia od obciążenia dla oleju z dodatkami (5% mas.) związków chlorowych (wg Winogradowej);
- - olej TS-14,5 + dwuchlorodwufenylotrojchloroetan (DDT),
- - olej TS-14,5 + ośmiochlorocyklopenten,
- - olej TS-14,5 + sześciochloroetan,
- - olej TS-14,5 + sowol,
- - olej TS-14,5 + chloroparafina
|
od alkilofosforanów. Możliwe jest również otrzymywanie dodatków fosforowych przez ogrzewanie mieszanin różnych związków organicznych z czerwonym fosforem.
- Spośród fosforanów najczęściej stosowanymi dodatkami smarnościowymi są: fosforan trójkrezylu, fosforan trójfenylu, fosforan trójtolylu itp. Stosowane są również propyle- i butylofosforany i fosforyny, np.
5.3.6.5. Związki siarkowo-chlorowe
Badania eksploatacyjne wykazały, że znacznie lepsze efekty smarnościowe uzyskuje się, gdy wprowadzane do oleju dodatki zawierają dwa lub więcej aktywnych pierwiastków. Przykładem takich dodatków mogą być dodatki zawierające siarkę i chlor. Dodatki te mogą składać się z dwóch składników, z których jeden jest związkiem siarkowym, a drugi chlorowym. Przykładem może być amerykański dodatek Santopoid A (mieszanina chloroparafiny i dwusiarczku dwubenzylu) lub radziecki dodatek EZ-5 (mieszanina siarkowanych terpenów i sześciochloroetanu). Możliwe jest również otrzymywanie dodatków siarkowochlorowych jednoskładnikowych, a więc zawierających chlor i siarkę w cząsteczce. Tego typu związki otrzymuje się zazwyczaj przez działanie chlorku lub dwuchlorku siarki na związki nienasycone.
- Istnieje wiele handlowych dodatków będących produktami działania chlorku siarki na tłuszcze roślinne, terpeny itp. Można je również otrzymać w reakcjach chlorowanych węglowodorów z alkilo- lub arylotiolami, siarczkami, ksantogenianami, trójtiowęglanami i merkaptanami metali alkalicznych. Również siarkowanie związków chloroorganicznych, jak i chlorowanie organicznych związków siarki prowadzi to tego celu.
5.3.6.6. Związki chlorowo-fosforowe
Związki chlorowo-fosforowe powodują wytworzenie na powierzchniach metali trwałej warstwy granicznej. Przykładem tego typu dodatku może być dodatek Bayer LG, który jest fosforanem chlorofenylowym. Jak podaje Winogradowa bardzo efektywnymi związkami tej grupy są estry kwasów chloroalkilofosfoniowych zawierających grupę CCl3, jak również estry chloroalkilowe kwasu fosforawego. Przykładem handlowego dodatku tego typu związków może być Chloref-40, będący estrem kwasu trójchlorometylofosfoniowego
W handlu znajduje się wiele znacznie bardziej złożonych, ale bardziej efektywnych dodatków chlorowo-fosforowych (rys. 5.76).
 |
Rys. 5.76.Zależność zużycia od obciążenia dla oleju z dodatkami (2% mas.) pochodnych kwasów chlorofosfonowych (wg Winogradowej);
- - olej TS-14,5 czysty,
- - chloref-40 (chem. czysty),
- - chloref-40 (techn.),
- - ester dwubutylowy kwasu jednochlorometylofosfonowego,
- - + chloref-amina,
- - ester dwuamylowy kwasu trójchloroacetoksyfosfonowego,
- - chloref-40 + dwualkilofosforan,
- - ester trójchloroamylowy kwasu metylofosfonowego
|
5.3.6.7. Związki siarkowo-fosforowe
Związki siarkowo-fosforowe są uważane za najbardziej uniwersalne dodatki smarnościowe. Wiąże się z tym bardzo duże ich praktyczne zastosowanie. Zwłaszcza dużą rolę odgrywają różnego rodzaju pochodne kwasu dwutiofosforowego i dwutiofosforawego oraz produkty reakcji pięciosiarczku fosforu z różnego rodzaju mieszaninami organicznymi, np. terpenami, estrami o dużej cząsteczce, tłuszczami. Możliwe jest również otrzymanie takich związków przez ogrzewanie różnych siarkowanych tłuszczów, olejów, polimerów niskocząsteczkowych z fosforem. Większość jednak dodatków zawierających fosfor i siarkę, to różnego rodzaju pochodne tiokwasów pięciowartościowego fosforu, jak:
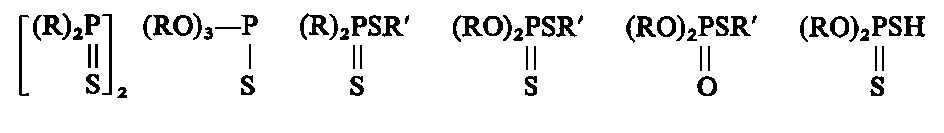
|
- Przez działanie kwasu dwualkilodwutiofosforowego na tlenki ziem alkalicznych można otrzymać ich sole
Powstający w wyniku tej reakcji dwualkilodwutiofosforan cynku jest powszechnie stosowany jako dodatek wielofunkcyjny, wykazujący działanie przeciwkorozyjne, przeciwutleniające i smarnościowe (krajowy dodatek Akorox-88). Sole baru i innych metali służą głównie jako dodatki przeciwutleniające i myjące. W alkilodwutiofosforanach cynku różne firmy stosują różnego rodzaju i długości podstawniki wzmagające efektywność działania tych związków (rys. 5.77). Należy zaznaczyć, że dla
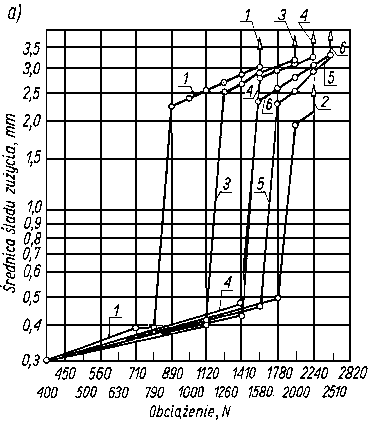
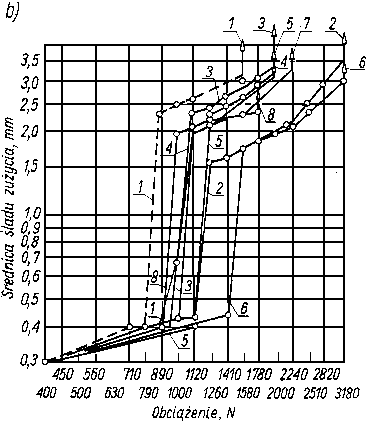 |
Rys. 5.77. Zależność zużycia od obciążenia dla oleju z dodatkiem (2% mas.) pochodnych kwasów (wg Winogradowej):
- a) fosforowych;
- - olej TS-14,5 czysty,
- - (C4H9O)2POH,
- - (C3H7O)3PO,
- - [C4H9O)2P(O)]2,
- - [C2H5O)2P(O)]2,
- - (C3H7O)2P(0)C3H7;
- b) tiofosforowych;
- - olej TS-14,5 czysty,
- - (C4H9S)3P,
- - (C5H11O)3PO,
- - (C5H11O)2P(O)SC5H11,
- - (C5H11O)2P(S)SC5H11,
- - (C5H11O)2P(S)SH,
- - (C3H7)2P(S)SC3H7,
- - [(C5H11O)2P(S)]2
|
zwiększenia trwałości warstwy olejowej stosuje się wiele innych, często bardzo złożonych związków fosforowo-siarkowych, których omówienie można znaleźć w literaturze o charakterze monograficznym.
5.3.6.8. Inne rodzaje dodatków smarnościowych
Literatura patentowa zawiera omówienie tysięcy różnych związków mogących znaleźć zastosowanie jako dodatki smarnościowe i ich pełne przedstawienie przekracza możliwości niniejszego opracowania. Wiele z tych związków zawiera po kilka aktywnych pierwiastków równocześnie (np. związki siarkowo-fosforowo-azotowe). Należy zaznaczyć, że ciekawym działaniem odznaczają się niektóre związki azotowe, a m.in. nitrowane oleje mineralne. Wzmagają one w dużym stopniu działanie innych dodatków. Przypuszczalnie należy to przypisać bardzo dużemu momentowi dipolowemu, jaki nadaje tym substancjom grupa nitrowa.
|